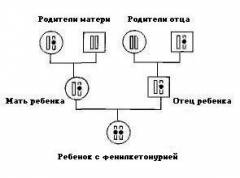
Фенилкетонурия (ФКУ) – заболевание, связанное с нарушением обмена веществ (b), – и альбинизм (а) наследуются у человека как рецессивные аутосомные несцепленные признаки. В семье отец – альбинос и болен ФКУ, а мать дигетерозиготна по этим генам. Составьте схему решения задачи, определите генотипы родителей, генотипы и фенотипы возможного потомства и вероятность рождения детей-альбиносов и ФКУ одновременно.
Какой закон наследования проявляется в данном случае?
Пояснение.
Схема решения задачи
Ответ:
1) генотипы родителей: мать – АаВb (гаметы AB, Ab, aB, ab),
отец – ааbb (гаметы ab);
2) генотипы и фенотипы возможного потомства:
АаВb – норма по двум парам признаков,
Ааbb – альбинизма нет, ФКУ,
ааВb – альбинизм, ФКУ отсутствует
ааbb – альбинизм, ФКУ;
3) 25% (ааbb) детей-альбиносов и больных ФКУ одновременно.
4) Проявляется закон независимого наследования признаков, так как гены не сцеплены и находятся в разных парах гомологичных хромосом
Фенилкетонурия (Болезнь Феллинга, Фенилпировиноградная олигофрения)

Фенилкетонурия – это наследственное нарушение аминокислотного обмена, обусловленное недостаточностью печеночных ферментов, участвующих в метаболизме фенилаланина до тирозина. Ранними признаками фенилкетонурии служат рвота, вялость или гиперактивность, запах плесени от мочи и кожи, задержка психомоторного развития; типичные поздние признаки включают олигофрению, отставание в физическом развитии, судороги, экзематозные изменения кожи и др. Скрининг новорожденных на фенилкетонурию проводится еще в родильном доме; последующая диагностика включает молекулярно-генетическое тестирование, определение концентрации фенилаланина в крови, биохимический анализ мочи, ЭЭГ, МРТ головного мозга. Лечение фенилкетонурии заключается в соблюдении специальной диеты.
Общие сведения
Фенилкетонурия (болезнь Феллинга, фенилпировиноградная олигофрения) – врожденная, генетически обусловленная патология, характеризующаяся нарушением гидроксилирования фенилаланина, накоплением аминокислоты и ее метаболитов в физиологических жидкостях и тканях с последующим тяжелым поражением ЦНС. Фенилкетонурия впервые описана А. Феллингом в 1934 г.; встречается с частотой 1 случай на 10 000 новорожденных.
В неонатальном периоде фенилкетонурия не имеет клинических проявлений, однако поступление фенилаланина с пищей вызывает манифестацию заболевания уже в первом полугодии жизни, а в дальнейшем приводит к тяжелым нарушениям развития ребенка. Именно поэтому пресимптоматическое выявление фенилкетонурии у новорожденных является важнейшей задачей неонатологии, педиатрии и генетики.

Фенилкетонурия
Причины фенилкетонурии
Фенилкетонурия является заболеванием с аутосомно-рецессивным характером наследования. Это означает, что для развития клинических признаков фенилкетонурии ребенок должен унаследовать по одной дефектной копии гена от обоих родителей, являющихся гетерозиготными носителями мутантного гена.
Чаще всего к развитию фенилкетонурии приводит мутация гена, кодирующего фермент фенилаланин-4-гидроксилазу и расположенного на длинном плече 12 хромосомы (локус12q22-q24.1). Это, так называемая, классическая фенилкетонурия I типа, составляющая 98% всех случаев заболевания. Гиперфенилаланинемия может достигать 30 мг% и выше. При отсутствии лечения данный вариант фенилкетонурии сопровождается глубокой умственной отсталостью.
Кроме классической формы, различают атипичные варианты фенилкетонурии, протекающие с той же клинической симптоматикой, но не поддающиеся коррекции диетотерапией. К ним относятся фенилкетонурия II типа (недостаточность дегидроптеринредуктазы), фенилкетонурия III типа (дефицит тетрагидробиоптерина) и другие, более редкие варианты. Вероятность рождения ребенка, больного фенилкетонурией, повышается при заключении близкородственных браков.
Патогенез
В основе классической формы фенилкетонурии лежит недостаточность фермента фенилаланин-4-гидроксилазы, участвующего в конверсии фенилаланина в тирозин в митохондриях гепатоцитов. В свою очередь, производный тирозина – тирамин является исходным продуктом для синтеза катехоламинов (адреналина и норадреналина), а дийодтирозин – для образования тироксина. Кроме этого, результатом метаболизма фенилаланина служит образование пигмента меланина.
Наследственная недостаточность фермента фенилалаиин-4-гидроксилазы при фенилкетонурии приводит к нарушению окисления фенилаланина, поступающего с пищей, в результате чего его концентрация в крови (фенилаланинемия) и спинномозговой жидкости значительно возрастает, а уровень тирозина соответственно падает. Избыточное содержание фенилаланина устраняется путем повышенной экскреции с мочой его метаболитов — фенилпировиноградной, фенилмолочной и фенилуксусной кислот.
Нарушение обмена аминокислот сопровождается нарушением миелинизации нервных волокон, снижением образования нейромедиаторов (дофамина, серотонина и др.), запускающими патогенетические механизмы задержки умственного развития и прогредиентное слабоумие.
Симптомы фенилкетонурии
Новорожденные с фенилкетонурией не имеют клинических признаков заболевания. Обычно манифестация фенилкетонурии у детей происходит в возрасте 2-6 месяцев. С началом кормления в организм ребенка начинает поступать белок грудного молока либо его заменителей, что приводит к развитию первых, неспецифических симптомов – вялости, иногда – беспокойства и гипервозбудимости, срыгивания, мышечной дистонии, судорожного синдрома. Одним из ранних патогномоничных признаков фенилкетонурии служит упорная рвота, которая нередко ошибочно расценивается как проявление пилоростеноза.
Ко второму полугодию становится заметным отставание ребенка в психомоторном развитии. Ребенок становится менее активным, безучастным, перестает узнавать близких, не пытается садиться и вставать на ножки. Аномальный состав мочи и пота обусловливают характерный «мышиный» запах (запах плесени), исходящий от тела. Часто наблюдается шелушение кожи, дерматиты, экзема, склеродермия.
У детей с фенилкетонурией, не получающих лечения, выявляется микроцефалия, прогнатия, позднее (после 1,5 лет) прорезывание зубов, гипоплазия эмали. Отмечается задержка речевого развития, а к 3-4 годам выявляется глубокая олигофрения (идиотия) и практически полное отсутствие речи.
Дети с фенилкетонурией имеют диспластическое телосложение, нередко — врожденные пороки сердца, вегетативные дисфункции (потливость, акроцианоз, артериальную гипотонию), страдают запорами. К фенотипическим особенностям детей, страдающих фенилкетонурией, следует отнести светлую кожу, глаза и волосы. Для ребенка с фенилкетонурией характерны специфическая поза «портного» (согнутые в суставах верхние и нижние конечности), тремор рук, шаткая, семенящая походка, гиперкинезы.
Клинические проявления фенилкетонурии II типа характеризуются тяжелой степенью умственной отсталости, повышенной возбудимостью, судорогами, спастическим тетрапарезом, сухожильной гиперрефлексией. Прогрессирование заболевание может приводить к гибели ребенка в возрасте 2-З лет. При фенилкетонури III типа развивается триада признаков: микроцефалия, олигофрения, спастический тетрапарез.
Диагностика
В настоящее время диагностика фенилкетонурии (а также галактоземии, врожденного гипотиреоза, адрено-генитального синдрома и муковисцидоза) входит в программу неонатального скрининга, осуществляемого всем новорожденным. Основные и дополнительные методы диагностики:
- Скрининг-тест. Проводится на 3-5 день жизни доношенного и 7 день жизни недоношенного ребенка путем забора образца капиллярной крови на специальный бумажный бланк. При обнаружении гиперфенилаланемии более 2,2 мг% ребенка направляют к детскому генетику для повторного обследования.
- Биохимические исследования. Для подтверждения диагноза фенилкетонурии проверяется концентрация фенилаланина и тирозина в крови, определяют активность печеночных ферментов (фенилаланингидроксилазы), выполняется биохимическое исследование мочи (определение кетоновых кислот), метаболитов катехоламинов в моче и др.
- Неврологическое обследование. Дополнительно проводится ЭЭГ и МРТ головного мозга, осмотр ребенка детским неврологом.
- Пренатальная диагностика. Генетический дефект при фенилкетонурии может быть обнаружен еще на этапе беременности в ходе инвазивной пренатальной диагностики плода (хорионбиопсии, амниоцентеза, кордоцентеза). В остальных случаях окончательный диагноз выставляется по результатам ДНК-диагностики после рождения.
Дифференциальный диагноз фенилкетонурии проводят с внутричерепной родовой травмой новорожденных, внутриутробными инфекциями, другими нарушениями обмена аминокислот.
Лечение фенилкетонурии
Основополагающим фактором в лечении фенилкетонурии является соблюдение диеты, ограничивающей поступление белка в организм. Лечение рекомендуется начинать при концентрации фенилаланина >6 мг%. Для грудных детей разработаны специальные смеси — Афенилак, Лофенилак; для детей старше 1 года – Тетрафен, Фенил-фри; старше 8 лет — Максамум-ХР и др. Основу диеты составляют низкобелковые продукты — фрукты, овощи, соки, белковые гидролизаты и аминокислотные смеси. Расширение диеты возможно после 18 лет в связи с возрастанием толерантности к фенилаланину. В соответствии с российским законодательством обеспечение лиц, страдающих фенилкетонурией, лечебным питанием, должна осуществляться бесплатно.
Больным назначается прием минеральных соединений, витаминов группы В и др.; по показаниям — ноотропные средства, антиконвульсанты. В комплексной терапии фенилкетонурии широко используется общий массаж, ЛФК, иглорефлексотерапия. Атипичные формы фенилкетонурии, не поддающиеся лечению диетой, требуют назначения гепатопротекторов, противосудорожных средств, заместительной терапии леводопой, 5-гидрокситриптофаном.
Дети, страдающие фенилкетонурией, находятся под наблюдением участкового педиатра и психоневролога; нередко нуждаются в помощи логопеда и дефектолога. Необходим тщательный мониторинг нервно-психического статуса детей, контроль уровня фенилаланина в крови и показателей электроэнцефалограммы.
Прогноз и профилактика
Проведения массового скрининга на фенилкетонурию в неонатальном периоде позволяет организовать раннюю диетотерапию и предотвратить тяжелые церебральные повреждения, нарушения функции печени. При раннем назначении элиминационной диеты при классической фенилкетонурии прогноз развития детей хороший. При поздно начатом лечении прогноз в отношении умственного развития неблагоприятный.
Профилактика осложнений фенилкетонурии заключается в проведении массового скрининга новорожденных, раннего назначения и длительного соблюдения диетического питания.
С целью оценки риска рождения ребенка с фенилкетонурией предварительное генетическое консультирование должны пройти супружеские пары, уже имеющие больного ребенка, состоящие в кровнородственном браке, имеющие родственников с данным заболеванием. Женщины с фенилкетонурией, планирующие беременность, должны соблюдать строгую диету до зачатия и во время беременности для исключения повышения уровня фенилаланина и его метаболитов и нарушения развития генетически здорового плода. Риск рождения ребенка с фенилкетонурией у родителей-носителей дефектного гена, составляет 1:4.
Фенилкетонурия — лечение в Москве
Сайт предоставляет справочную информацию. Адекватная диагностика и лечение болезни возможны под наблюдением добросовестного врача. У любых препаратов есть противопоказания. Необходима консультация специалиста, а также подробное изучение инструкции!
Фенилкетонурия (ФКУ) – довольно редкое наследственное заболевание, связанное с нарушением обмена аминокислот. Организм больного фенилкетонурией человека не способен расщеплять аминокислоту фенилаланин, которая поступает с белковой пищей. В результате этого, в тканях накапливаются соединения, отравляющие нервную систему и головной мозг в частности. Развивается умственная отсталость (малоумие), вплоть до идиотии. В связи с этим болезнь получила и другое название – фенилпировиноградная олигофрения.
Однако из всех наследственных заболеваний фенилкетонурия, единственное, которое удается полностью нейтрализовать. Сегодня ребенка, рожденного с признаками ФКУ, можно вырастить абсолютно здоровым. Обезопасить мозг малыша удается с помощью специальной диеты, о которой мы расскажем ниже.
В разных странах частота этого заболевания отличается в разы. В России рождается один больной ребенок на 10 000. В некоторых регионах Великобритании этот показатель в два раза выше – 1:5000. Дети на Африканском континенте практически не болеют фенилкетонурией. Среди больных количество девочек почти в два раза превышает количество мальчиков.
Механизм развития заболевания
 Заболевание наследуется только в том случае, если оба родителя передали ребенку склонность к болезни, и поэтому встречается довольно редко. У двух процентов людей есть измененный ген, который отвечает за развитие болезни. При этом человек остается полностью здоровым. Но когда мужчина и женщина, носители мутировавшего гена, вступают в брак и решают завести детей, то вероятность того, что малыши будут страдать от фенилкетонурии, составляет 25%. А возможность того, что дети будут носителями патологического гена ФКУ, но сами останутся практически здоровыми, составляет 50%.
Заболевание наследуется только в том случае, если оба родителя передали ребенку склонность к болезни, и поэтому встречается довольно редко. У двух процентов людей есть измененный ген, который отвечает за развитие болезни. При этом человек остается полностью здоровым. Но когда мужчина и женщина, носители мутировавшего гена, вступают в брак и решают завести детей, то вероятность того, что малыши будут страдать от фенилкетонурии, составляет 25%. А возможность того, что дети будут носителями патологического гена ФКУ, но сами останутся практически здоровыми, составляет 50%.
Причина возникновения этого заболевания связана с тем, что в печени человека не вырабатывается особый фермент – фенилаланин-4-гидроксилаза. Он отвечает за превращение фенилаланина в тирозин. Последний входит в состав пигмента меланина, ферментов, гормонов и необходим для нормальной работы организма.
При ФКУ фенилаланин, в результате побочных путей обмена, превращается в вещества, которых не должно быть в организме: фенилпировиноградную и фенилмолочную кислоты, фенилэтиламин и ортофенилацетат. Эти соединения накапливаются в крови и оказывают комплексное действие:
- нарушают процессы жирового обмена в мозге
- вызывают дефицит нейромедиаторов, которые передают нервный импульс между клетками нервной системы
- оказывают токсическое действие, отравляя мозг
Это вызывает значительное и необратимое снижение интеллекта. У ребенка быстро развивается умственная отсталость – олигофрения.
Симптомы фенилкетонурии
 Дети с ФКУ рождаются абсолютно здоровыми. Поэтому, если в течение первых дней жизни выявить заболевание и придерживаться диеты, то удается предотвратить разрушение мозга ребенка. При этом, никакие признаки заболевания не появляются. Малыш развивается и растет, как и его сверстники.
Дети с ФКУ рождаются абсолютно здоровыми. Поэтому, если в течение первых дней жизни выявить заболевание и придерживаться диеты, то удается предотвратить разрушение мозга ребенка. При этом, никакие признаки заболевания не появляются. Малыш развивается и растет, как и его сверстники.
Если же момент упущен, и ребенок употребляет в пищу белковые продукты, богатые фенилаланином, то начинают проявляться симптомы поражения центральной нервной системы. Поначалу изменения у больных фенилкетонурией незначительны. Их трудно заметить даже опытному педиатру. Это слабость и беспокойство. Малыш не улыбается и мало двигается.
К шести месяцам задержка развития становится более заметной. Ребенок слабо реагирует на происходящее, не узнает мать, не пытается сесть и перевернуться. Фенилаланин и его производные выводятся из организма с мочой и потом. Они вызывают специфический «мышиный» или затхлый запах.
Годовалый ребенок не умеет выражать голосом свои эмоции и переживания, имеет невыразительную мимику, не понимает речь родителей.
В возрасте трех лет и старше симптомы фенилкетонурии нарастают. У детей наблюдается повышенная возбудимость, утомляемость, нарушения поведения, психотические расстройства, умственная отсталость. Если не заниматься лечением фенилкетонурии, то состояние больного будет ухудшаться.
Диагностика фенилкетонурии
 В том случае, если есть подозрение, что один или оба родителя являются носителями гена ФКУ, то определить это можно в федеральных медико-генетических центрах. Для установления этого факта проводится генетическая экспертиза.
В том случае, если есть подозрение, что один или оба родителя являются носителями гена ФКУ, то определить это можно в федеральных медико-генетических центрах. Для установления этого факта проводится генетическая экспертиза.
На сегодняшний день все новорожденные дети массово обследуются на наличие фенилкетонурии. На территории России этот вопрос регламентирует приказ Минздрава РФ №316 от 30.12.1993 г. Процедура получила название неонатальный скрининг и является эффективным способом выявления наиболее распространенных наследственных заболеваний, среди них и ФКУ.
Массовое обследование новорождённых это простой и достоверный метод диагностики. В роддоме у каждого ребенка берут несколько капель периферической крови из пяточки. Это делается натощак, через три часа после кормления. У доношенных детей анализ берут на четвертый день жизни, а у недоношенных на седьмой. У тех новорожденных, которые появились на свет не в родильных домах, важно взять анализ на протяжении первых трех недель.
Кровь наносят на специальный тест-бланк, который потом отправляют в лабораторию для проведения генетического исследования. Там на протяжении суток проводится анализ крови на содержание в ней аминокислоты — фенилаланина. Результаты теста заносятся в обменную карту ребенка в виде штампа: «На ФКУ и ВГ обследован».
В том случае, если в анализе обнаруживают измененный ген, то родителей с ребенком приглашают в медико-генетический центр для обследования. Для того, чтобы подтвердить или опровергнуть диагноз назначаются дополнительные исследования:
- в сухом пятне крови
- в сыворотке крови
- потовый тест
- копрограмма
- ДНК-диагностика
В любом случае родители должны понимать, что при своевременно начатом лечении и соблюдении диеты удается полностью предотвратить развитие болезни.
Лечение фенилкетонурии
На сегодняшний день в нашей стране единственным эффективным методом лечения является диетотерапия. Разрабатываются препараты, которые позволят контролировать уровень фенилаланина в крови без соблюдения диеты. В этом направлении есть значительные успехи, но в продаже такие лекарственные средства появятся не раньше чем через 5-7 лет.
Постоянно идет работа над поиском новых средств и методов борьбы с болезнью.
- Перспективным направлением считается использование растительного фермента фенилаланинлиазы, который будет расщеплять излишки фенилаланина в организме.
- Ученые возлагают большие надежды на генотерапию с использованием вирусного фактора, которая позволит вылечить больной ген и полностью избавиться от проблемы.
- Практикуется введение гена фенилаланингидроксилазы прямо в пораженные клетки печени.
Но в нашей стране эти разработки пока не используются. Лечение с помощью диеты – основная помощь больным фенилкетонурией. Ограничить употребление белка необходимо с самого рождения и до половой зрелости. За ростом и развитием ребенка постоянно наблюдают врачи: педиатр и невролог. Специалисты корректируют количество белков, чтобы они соответствовали возрасту и нагрузкам ребенка.
Некоторые формы фенилкетонурии поддаются лечению тетрагидробиоптерином, который является составляющей частью недостающего фермента фенилаланин-4-гидроксилазы. Атипичные формы ФКУ не лечатся с помощью диеты и требуют регулярного приема тетерагидробиоптерина или его заменителей.
Питание больного фенилкетонурией
 Для того, чтобы нервные клетки ребенка не подвергались токсическому воздействию фенилаланина и его производных, нужно полностью исключить из рациона животные белки. Если это сделать на первых неделях жизни, то мозг останется полностью здоровым. Если же начинать ограничивать белок в более позднем возрасте, то задержку развития удается несколько приостановить. Но вернуть здоровье нервной системе и устранить изменения в нервных клетках уже не удастся.
Для того, чтобы нервные клетки ребенка не подвергались токсическому воздействию фенилаланина и его производных, нужно полностью исключить из рациона животные белки. Если это сделать на первых неделях жизни, то мозг останется полностью здоровым. Если же начинать ограничивать белок в более позднем возрасте, то задержку развития удается несколько приостановить. Но вернуть здоровье нервной системе и устранить изменения в нервных клетках уже не удастся.
Соблюдать диету требуется до 16-18 лет. Это обязательное условие. Желательно контролировать количество животных белков и в дальнейшем.
Если женщина, у которой в детстве были обнаружены признаки ФКУ, планирует забеременеть, то ей обязательно нужно вернуться к диете без фенилаланина. Таких ограничений необходимо придерживаться до зачатия, во время беременности и кормления грудью.
Все необходимые для роста и развития аминокислоты поступают в организм из специализированных лечебных продуктов. Обычно они представляют собой порошок – сухую смесь аминокислот. Их родителям больного малыша выдают бесплатно в медико-генетической консультации.
Грудные дети получают специальные смеси полностью очищенные от лактозы на основе гидролизата молочного белка.
Питательные средства, которые должны заменить детям натуральные белковые продукты, содержат:
- пептиды (расщепленные ферментами молочные белки);
- свободные аминокислоты (тирозин, триптофан, цистин, гистидин и таурин).
В России применяются такие смеси: Афенилак, Аналог-СП, Мдмил-ФКУ-0. Они представляют собой порошки, которые необходимо разводить кипяченой водой или сцеженным грудным молоком, согласно инструкции. В результате получается жидкая смесь или «сметанка». Такой прикорм вводят постепенно, в течение 2-5 дней под наблюдением врача.
Специальными диетическими продуктами для пополнения запасов белка у детей разного возраста также являются: “Берлафен“, “Циморган“, “Минафен“, “Апонти“.
Детей, больных ФКУ, можно кормить грудью. Но при этом кормящей матери нужно придерживаться специальной диеты.
В диете детей дошкольного и школьного возраста полностью исключают из меню белковые продукты. В списке разрешенных продуктов – овощи, фрукты, изделия из крахмала, растительные масла. При составлении дневного меню необходимо строго придерживаться возрастных норм фенилаланина.
| Возраст ребенка | Суточное количество фенилаланина (мк/кг массы тела) |
| Младше 2 мес. | 60 |
| 2-3 мес. | 60-55 |
| 3-6 мес. | 55-45 |
| 6-12 мес. | 45-35 |
| 1-1,5 года | 35-30 |
| 1,5-3 года | 30-25 |
| 3-6 лет | 25-15 |
| Старше 6 лет | 15-10 |
Необходимо помнить, что для растущего организма полноценное питание жизненно необходимо. Так в сутки ребенку требуется 120 мг тирозина на каждый килограмм массы. Поэтому дети и подростки с таким диагнозом должны получать аминокислоты для построения клеток и роста из дополнительных источников. Также обязательно назначают витаминно-минеральный комплекс. Особенно важно, чтобы ребенок получал норму витаминов С, В6 и B1, фолиевой кислоты, железа, кальция и магния. Количество калорий должно быть увеличено на 30% по сравнению с дневной нормой сверстников.
Группы продуктов при ФКУ
Разделяют три группы натуральных продуктов. В основе классификации лежит количество в них фенилаланина:
- Красный список – продукты, которые необходимо полностью исключить из рациона.
- Оранжевый список – разрешены в небольших количествах под строгим контролем.
- Зеленый список – могут употребляться без ограничений.
| Красный список | Оранжевый список | Зеленый список |
| Все виды мяса | Молочные продукты | Фрукты |
| Колбасные изделия | Рис и кукуруза | Ягоды |
| Все виды рыбы | Овощи (картофель, капуста) | Зелень |
| Морепродукты | Овощные консервы | Овощи |
| Яйца | Рисовая, кукурузная мука | |
| Сыры | Крахмал и саго | |
| Творог | Сахар и варенье | |
| Орехи | Мед | |
| Хлеб и хлебобулочные изделия | Сливочное и растительное масло, топленый жир | |
| Кондитерские изделия | ||
| Крупы и хлопья | ||
| Продукты из сои | ||
| Поп-корн | ||
| Аспартам |
Промышленность выпускает еще две группы продуктов:
- искусственные низкобелковые продукты, специально для диетического питания (хлеб, печенье, макароны)
- готовые пюре для детского питания на основе фруктов.
На основе этих продуктов можно составить полноценное меню, готовить ребенку вкусные, полезные и разнообразные блюда.
Родителям ребенка, больного ФКУ, важно уметь составлять диету и правильно рассчитывать количество фенилаланина. Для этого необходимо иметь под рукой весы, которые дают возможность взвешивать до десятой доли грамма.
Контроль уровня фенилаланина в крови
 Необходимо контролировать количество фенилаланина. Оно должно находиться в границах 3–4 мг% или 180–240 мкмоль/л.
Необходимо контролировать количество фенилаланина. Оно должно находиться в границах 3–4 мг% или 180–240 мкмоль/л.
Для определения необходимо сделать анализ крови в лаборатории. До трехмесячного возраста это делают еженедельно.
Постепенно врач снижает количество анализов. С тех месяцев до года – один раз в месяц, с года до трех лет – один раз в два месяца. После трех лет частота проверок снижается до одного раза в три месяца. Существует специальная схема, но специалист может изменить ее, исходя из состояния больного.
Делать анализ желательно утром натощак. От качества и регулярности такого контроля и своевременных исправлений в диете зависит сохранение интеллекта.
Ответы на часто задаваемые вопросы
Как проявляется фенилкетонурия у новорожденных?
Новорожденные с диагнозом ФКУ ничем не отличаются от здоровых детей. И если болезнь вовремя выявить и остановить ее развитие, то и в дальнейшем такой ребенок останется абсолютно здоровым.
Как выглядят больные фенилкетонурией? 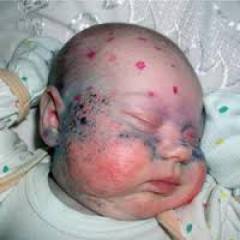
При рождении младенцы, больные фенилкетонурией, ничем не отличаются от остальных детей. Но на втором месяце жизни начинают проявляться изменения:
- посветление волос и радужки глаза из-за недостатка пигмента меланина
- чрезмерная прибавка в весе
- быстро зарастает большой родничок
- суховатая кожа
- шелушение, сыпь и экзема
- частая рвота
- моча и пот с характерным «мышиным» запахом
На втором полугодье дети, не получающие лечение, перестают узнавать мать, не могут фиксировать взгляд на одном предмете, не реагируют на яркие игрушки, не садятся и не переворачиваются, становятся раздражительными. В возрасте 2-3 года отмечаются такие особенности:
- появляются судороги и спазмы
- скованность движений и зажатая «поза портного», что связанно с повышенным напряжением в мышцах
- неадекватное поведение, выкрики, смех
- уменьшение размеров черепа
- деформация ушных раковин
- дрожание пальцев рук
- недержание мочи
- выступающая вперед нижняя челюсть
Внешние признаки болезни выражены незначительно, но при отсутствии диеты развиваются сильные психические отклонения, приводящие к инвалидности.
Какие смеси использовать для ребенка с фенилкетонурией?
Для того чтобы обеспечить детей всеми необходимыми веществами в достаточном количестве разработаны специальные смеси на основе незаменимых и заменимых аминокислот. В их состав также входят витамины и все необходимые микроэлементы.
Для детей до одного года рекомендуют:
- Афенилак 13, Афенилак 15 от компании «Нутритек», Россия;
- MIDмил ФКУ 0 (Hero, Испания);
- ХР Аналог («Нутриция», Голландия);
- Фенил Фри 1 («Мид Джонсон» США).
Для детей старше одного года и для взрослых:
- П-АМ 1, П-АМ 2, П-АМ 3;
- Изифен (готовый продукт), а также ХР Максамейд и ХР Максамум с нейтральным и фруктовым вкусами («Нутриция», Голландия).
Эти продукты имеют прекрасные вкусовые качества и хорошо переносятся. Они необходимы детям и взрослым с диагнозом ФКУ в периоды умственных и физических нагрузок. Смеси удобны в применении, питательны и полностью покрывают потребности организма в аминокислотах.
Какова продолжительность жизни больного с фенилкетонурей?
Если человеку вовремя было назначено соответствующее лечение, то продолжительность и качество его жизни никак не отличается от остальных членов общества. В том случае, если развилось слабоумие, то продолжительность жизни резко сокращается.
Как лечить фенилкетонурию?
 На сегодняшний день в России для лечения ФКУ используют специальную безфенилаланиновую диету. Для пополнения запасов тирозина и других аминокислот, все больные дети бесплатно получают специальные препараты. Соблюдать диету желательно до 18 лет, хотя ряд врачей утверждает, что лучше делать это на протяжении всей жизни.
На сегодняшний день в России для лечения ФКУ используют специальную безфенилаланиновую диету. Для пополнения запасов тирозина и других аминокислот, все больные дети бесплатно получают специальные препараты. Соблюдать диету желательно до 18 лет, хотя ряд врачей утверждает, что лучше делать это на протяжении всей жизни.
Такой метод диетотерапии является самым дешевым и действенным. На протяжении многих лет он помогает детям с этой болезнью вырасти здоровыми. В своем развитии они ничем не уступают сверстникам. Те, кому в детстве ставили диагноз «фенилкетонурия», учатся в школе, получают высшее образование, заводят семью и рожают здоровых детей.
Подводя итоги, отметим, что фенилкетонурия это тяжелое генетическое заболевание, которое может привести к психической инвалидности. Изменения происходят в нервной системе очень быстро и имеют необратимый характер. Однако можно не допустить развития болезни. Для этого необходима ранняя диагностика и специальная диета.
Какие бывают типы фенилкетонурии?
Выделяют 3 типа фенилкетонурии:
- Фенилкетонурия I. Классическая и наиболее распространенная форма заболевания, описанная выше в статье. Связана с мутацией гена в 12-й хромосоме, при этом нарушается образование фермента фенилаланин-4-гидроксилазы, который превращает фенилаланин в тирозин.
- Фенилкетонурия II. При этой форме заболевания нарушение происходит в 4-й хромосоме. Нарушается выработка фермента дигидроптеридинредуктазы, который также способствует превращению фенилаланина в тирозин. Заболевание наследуется так же, как и I форма: для того, чтобы родился больной ребенок, необходимо, чтобы носителями гена были оба родителя. Распространенность фенилкетонурии II – 1 случай на 100 000 новорожденных.
- Фенилкетонурия III. В результате генетических нарушений возникает недостаток фермента 6-пирувоилтетрагидроптеринсинтазы. Наследуется, как и две предыдущие формы заболевания. Распространенность – 1 случай на 300 000 новорожденных.
Дают ли инвалидность при фенилкетонурии?
Критерии установления инвалидности при фенилкетонурии:
- При фенилкетонурии I инвалидность устанавливают только при необратимых нарушениях со стороны центральной нервной системы, которые приводят к неврологическим расстройствам и умственной отсталости.
- При фенилкетонурии II и III типа группу инвалидности устанавливают во всех случаях.
Существует ли профилактика фенилкетонурии?
 Специальной профилактики фенилкетонурии не существует. Но некоторые мероприятия помогают правильно оценить риски, вовремя принять необходимые меры:
Специальной профилактики фенилкетонурии не существует. Но некоторые мероприятия помогают правильно оценить риски, вовремя принять необходимые меры:
- Генетическое консультирование. Необходимо людям, планирующим завести ребенка, которые больны или являются носителями неправильного гена, у которых болен хотя бы один близкий родственник или уже родился больной ребенок. Консультирование проводит врач-генетик. Он помогает разобраться, как ген, ответственный за фенилкетонурию, передавался в предыдущих поколениях, каковы риски будущего ребенка. Также генетик помогает с планированием семьи.
- Скрининг новорожденных. Анализ не помогает предотвратить заболевание, но позволяет выявить его максимально рано, пока оно еще не привело к необратимым изменениям в головном мозге.
- Консультации и диета для женщин, страдающих фенилкетонурией. Если вы женщина и страдаете ФКУ, вам следует проконсультироваться с врачом и спросить, когда лучше планировать беременность в вашем случае. Во время беременности нужно соблюдать правильную диету – это помогает предотвратить дефекты развития у ребенка.
Каков прогноз при фенилкетонурии?
Прогноз зависит от формы заболевания и начала лечения, соблюдения диетических рекомендаций, медико-педагогической коррекции.
При фенилкетонурии I, при своевременном начале необходимых мероприятий, прогноз, как правило, благоприятный. Ребенок растет и развивается нормально. Если опоздать с лечением и диетой, то результат будет не таким хорошим.
При фенилкетонурии II и III прогноз серьезнее. Соблюдение диеты не приносит эффекта.
Каковы факторы риска фенилкетонурии?
- Как уже упоминалось в статье, ребенок рискует получить заболевание или стать носителем мутантного гена, если он есть у обоих родителей.
- Среди разных этнических групп распространенность фенилкетонурии различается. Например, среди представителей негроидной расы неправильный ген встречается реже.
- В группе повышенного риска находятся дети матерей, страдающих фенилкетонурией. Если во время беременности женщина не придерживается специальной диеты, у ребенка могут возникать дефекты развития.
Полезные советы для больных фенилкетонурией

- Постоянно осуществляйте тщательный контроль. Если вам или вашему ребенку нужно придерживаться диеты с низким содержанием фенилаланина, необходимо ежедневно вести учет съеденной пищи.
- Постарайтесь, чтобы расчеты были максимально точными. Используйте специальные мерные стаканчики, ложки, весы, которые могут измерять массу в граммах. Это поможет четко контролировать количество съеденного фенилаланина каждый день.
- Используйте для учета количества фенилаланина в вашем рационе или в рационе вашего ребенка пищевой дневник или специальную компьютерную программу.
- Не обязательно сильно ограничивать себя. Сегодня можно купить специальные продукты с низким содержанием фенилаланина, например, макароны, рис, муку, хлеб, и питаться почти как обычный человек.
- Будьте изобретательны. Поговорите со своим лечащим врачом, диетологом: возможно, они посоветуют вам, как можно разнообразить рацион питания, не жертвуя здоровьем. Вы можете сделать свои блюда более разнообразными при помощи приправ.
Фенилкетонурия: причины появления, симптомы, диагностика и способы лечения.
Определение
Фенилкетонурия (ФКУ) – группа аутосомно-рецессивных заболеваний, обусловленных нарушением обмена незаменимой аминокислоты фенилаланина, поступающей в организм человека с белковой пищей.
 Причины появления фенилкетонурии
Причины появления фенилкетонурии
Причина заболевания связана с нарушением обмена незаменимой аминокислоты фенилаланина, приводящим к повышению ее уровня в крови, тканях и биологических жидкостях.
Избыток фенилаланина токсичен для нервной системы и при длительном воздействии вызывает в ней необратимые дегенеративные изменения.
Фенилаланин не синтезируется в клетках организма и поступает только с пищей. Метаболизм этой аминокислоты осуществляется двумя путями: участвует в биосинтезе белка или превращается в тирозин под действием фермента фенилаланингидроксилазы. В клетках меланоцитах, которые присутствуют в коже, волосах, сетчатке глаза, тирозин превращается в пигмент меланин. В щитовидной железе из тирозина синтезируются гормоны тироксин и трийодтиронин. В мозговом веществе надпочечников и нервной ткани тирозин является предшественником катехоламинов – дофамина, норадреналина, адреналина.
Классификация заболевания
Фенилкетонурия объединяет несколько форм нарушения обмена фенилаланина, сходных по клиническим признакам:
- классическая (ФКУ I типа), обусловленная мутацией гена ФАГ (РАН) фенилаланингидроксилазы;
- ФКУ II типа, обусловленная мутацией структурного гена для цитозольной дигидроптеридинредуктазы, что вызывает метаболический блок на путях превращения фенилаланина в тирозин, а также предшественников нейромедиаторов катехоламинового и серотонинового ряда;
- ФКУ III типа вызвана мутацией структурного гена для цитозольной пирувоилтетрагидроптерин синтазы, что приводит к ее недостаточности в печени и эритроцитах.
Тип наследования всех форм заболевания – аутосомно-рецессивный.
По уровню фенилаланина в сыворотке крови выделяют:
- легкую гиперфенилаланинемию – 120-600 мкмоль/л (2−10 мг/дл);
- умеренную (мягкую, среднюю) ФКУ – 600-1200 мкмоль/л (10−20 мг/дл);
- классическую (тяжелую) ФКУ – 1200 мкмоль/л и более (>20 мг/дл).
Симптомы фенилкетонурии
Новорожденный с ФКУ выглядит здоровым, а первые симптомы манифестируют при употреблении в пищу белковой пищи, содержащей большое количество фенилаланина.
Проявления заболевания зависят от тяжести мутационного повреждения фермента, степени активности фермента и сроков начала терапии.
Классическая фенилкетонурия характеризуется полным или почти полным дефицитом фенилаланингидроксилазы в печени. Проявляется у ребенка в возрасте 2-6 месяцев вялостью, отсутствием интереса к окружающему, иногда повышенным беспокойством, срыгиванием, снижением мышечного тонуса, признаками атопического дерматита, задержкой психомоторного развития, судорогами.
Эпилептические приступы могут быть первым признаком болезни и встречаются почти у половины нелеченных больных. Приступы носят упорный характер и плохо поддаются терапии.
Во втором полугодии жизни дети перестают реагировать на обращенную к ним речь, узнавать родителей, не фиксируют взгляд и не реагируют на яркие игрушки, не переворачиваются на живот, не сидят. Отмечается задержка статико-моторного и психоречевого развития.
Нарушения обмена аминокислот приводят к гипопигментации волос и кожи, поскольку у детей почти не вырабатывается пигмент меланин. В результате присутствует гиперчувствительность к инсоляции, может наблюдаться тяжелая экзема, дерматит, фолликулярный кератоз, повышенная склонность к гнойничковым инфекциям. У больных отмечают «мышиный» запах мочи, который объясняется выделением метаболитов фенилаланина. При отсутствии лечения медленно прогрессирует умственная отсталость. Могут наблюдаться двигательные, психопатоподобные и шизофреноподобные расстройства.
Умеренная фенилкетонурия характеризуется низкой остаточной ферментативной активностью. Клинически болезнь проявляется на первом-втором году жизни и медленно прогрессирует при отсутствии лечения.
При легкой форме заболевания клинические признаки отсутствуют либо выражены слабо.
Клинические проявления ФКУ II сходны с классической фенилкетонурией. Однако есть и свои особенности: гибель нейронов, кальцификация и анормальная васкуляризация центральной коры, белого вещества, базальных ганглий и таламуса, а также нарушение метаболизма фолатов. В клинической картине преобладает тяжелая умственная отсталость, судороги, признаки повышенной возбудимости, мышечная дистония (непроизвольные движения и формирование патологических поз), спастический тетрапарез (комплекс двигательных нарушений верхних и нижних конечностей).
Течение болезни прогрессирующее и нередко приводит к смерти в возрасте 2-3 лет.
Проявления фенилкетонурии III напоминают болезнь Паркинсона. Наблюдаются затруднения в удержании равновесия в определенной позе или при смене позы, недостаточная двигательная активность с ограничением скорости и объема движений, увеличение секреции слюнных желез, нарушения глотания, а также тяжелая умственная отсталость, микроцефалия.
Игнорирование рекомендаций по диетотерапии, недостаточный контроль уровня фенилаланина в крови могут иметь отдаленные последствия: низкий коэффициент интеллекта, замедленная речь, нарушения памяти, проблемы с концентрацией внимания и поведением.
У взрослых пациентов, прекративших соблюдение диеты, наблюдается ухудшение неврологического и психологического состояния, возникает поздняя эпилепсия, атаксия, тремор, депрессивные расстройства, неврозы.
Если пациенты соблюдают диету с ограничением высокобелковых продуктов, но не принимают специальные аминокислотные смеси без фенилаланина, существует риск развития дефицита витаминов, микро- и макроэлементов.
Женщинам с ФКУ до зачатия и во время беременности необходимо строго придерживаться рекомендованного врачом диетического питания, в противном случае у родившихся детей может диагностироваться синдром материнской фенилкетонурии, который проявляется дисморфией лица, задержкой умственного и физического развития, микроцефалией (значительным уменьшением размера черепа), врожденными пороками сердца.
 Диагностика фенилкетонурии
Диагностика фенилкетонурии
Определить риск рождения ребенка с ФКУ можно с помощью генетической экспертизы у обоих родителей до зачатия ребенка:
- фенилкетонурия, PAH м.;
Фенилкетонурия, PAH м.
Исследование мутаций в гене PAH.
Фенилкетонурия (гиперфенилаланинемия, ФКУ, ГФА, OMIM261600) — группа гетерогенных аутосомно-рецессивных заболеваний, обусловл�…
Фенилкетонурия, PAH ч.м.
Исследование частых мутаций в гене PAH.
Фенилкетонурия (гиперфенилаланинемия, ФКУ, ГФА, OMIM261600) — группа гетерогенных аутосомно-рецессивных заболеваний, об…
Основные наследственные заболевания
Определение носительства частых мутаций в генах, ответственных за развитие наиболее частых аутосомно-рецессивных заболеваний: муковисцидоз, несиндромальная ней�…
Главным критерием диагностики ФКУ является повышенное содержание фенилаланина в крови. Все формы заболевания можно диагностировать уже в первые дни жизни ребенка – для этого проводят биохимический скрининг новорожденных на наличие гиперфенилаланинемии.
В родильном доме на 4-й день жизни (у недоношенных детей на 7-й день) берут кровь из пятки. Значение фенилаланина выше 2,0 мг/дл классифицируется как гиперфенилаланинемия и требует проведения уточняющей диагностики. Ребенка наблюдают в медико-генетической консультации в течение первого года жизни с ежемесячным контролем уровня фенилаланина крови. При его концентрации выше 8,0 мг/дл диагностируется фенилкетонурия и назначается диетотерапия.
- ЭКГ и ЭхоКГ могут быть назначены для своевременного выявления патологии сердечно-сосудистой системы.
Эхокардиография
Исследование, позволяющее оценить функциональные и органические изменения сердца, его сократимость, а также состояние клапанного аппарата.
МРТ головного мозга
Безопасное и информативное сканирование структур головного мозга для диагностики его патологий.
- клинический анализ крови: общий анализ, лейкоформула, СОЭ (с микроскопией мазка крови при наличии патологических сдвигов);
- биохимический анализ крови, включающий: общий белок, альбумин (в крови), белковые фракции; оценку показателей работы почек (мочевина, креатинин, клубочковая фильтрация); оценку показателей работы печени (билирубин, АЛТ, АСТ); оценку углеводного обмена: глюкоза (в крови), глюкозотолерантный тест с определением глюкозы в венозной крови натощак и после нагрузки через 2 часа.
Общий белок (в крови) (Protein total)
Синонимы: Общий белок сыворотки крови; Общий сывороточный белок.
Total Protein; Serum Тotal Protein; Total Serum Protein; TProt; ТР.
Краткая характеристика определяемого вещества Общий бе�…
Альбумин (в крови) (Albumin)
Синонимы: Человеческий сывороточный альбумин; ЧСА; Альбумин плазмы;
Human Serum Albumin; ALB.
Краткая характеристика исследуемого вещества Альбумин
Альбумин – эт…
Креатинин (в крови) (Creatinine)
Синонимы: Анализ крови на креатинин; Сывороточный креатинин; Креатинин сыворотки, оценка СКФ. Сreat; Сre; Blood Creatinine; Serum Creatinine; Serum Creat.
Краткая характеристика определя�…
Мочевина (в крови) (Urea)
Синонимы: Диамид угольной кислоты; Карбамид; Мочевина в крови; Азот мочевины.
Urea nitrogen; Urea; Blood Urea Nitrogen (BUN); Urea; Plasma Urea.
Краткая характеристика аналита Мочевина
Моче�…
Глюкоза (в крови) (Glucose)
Материал для исследования
Сыворотка или плазма крови. Если нет возможности центрифугировать пробу через 30 минут после взятия для отделения сыворотки/плазм…
- Диагностика нарушений липидного обмена:
- триглицериды;
Триглицериды (Triglycerides)
Синонимы: Липиды крови; нейтральные жиры; ТГ.
Triglycerides; Trig; TG.
Краткая характеристика определяемого вещества Триглицериды
Триглицериды (ТГ) – источник получен…
- холестерин общий;
- холестерин-ЛПВП;
- холестерин-ЛПНП;
- Диагностика состояний дефицита:
- фолиевая кислота;
Фолиевая кислота (Folic Acid)
Фолиевая кислота − витамин, необходимый для нормального синтеза ДНК (особенно в онтогенезе) и процессов кроветворения.
Синонимы: Витамин В9; Фолацин; Пте�…
- витамин B12 (цианокобаламин);
- витамин А в сыворотке (ретинол);
- 25-OH витамин D;
- жиро- и водорастворимые витамины;
- жирорастворимые витамины;
- водорастворимые витамины;
- витамин B6, пиридоксаль-5-фосфат, плазма;
- витамин В1 – тиамин, плазма;
- витамин В2 – рибофлавин, плазма;
- витамин В5 – пантотеновая кислота.
К каким врачам обращаться
Пациентам с установленным диагнозом фенилкетонурии рекомендованы:
- прием врача-генетика с целью консультирования, назначения и коррекции диетотерапии;
- врача-педиатра детям с установленным диагнозом каждые 6 месяцев для оценки общего состояния здоровья и определения тактики дальнейшей терапии;
- врача-терапевта пациентам старше 18 лет с установленным диагнозом каждые 6 месяцев для оценки общего состояния здоровья и определения тактики дальнейшей терапии;
- врача-кардиолога при изменениях на электрокардиограмме;
- врача-невролога для своевременного выявления и мониторинга неврологических изменений;
- врача-офтальмолога с целью выявления глазной патологии;
- врача-гастроэнтеролога с целью выявления гастроэнтерологических нарушений;
- врача-психиатра для оценки психического статуса;
- врача-эндокринолога с целью уточнения нарушения функции эндокринной системы;
- акушера-гинеколога и врача-генетика во время беременности с целью коррекции терапии и контроля состояния плода;
- врача-диетолога для дополнительной коррекции диетотерапии.
Лечение фенилкетонурии
Основная цель лечения – снизить уровень фенилаланина в крови, повысить переносимость фенилаланина, получаемого с пищей.
Заболевание, протекающее в легкой форме, подлежит наблюдению и проведению дифференциальной диагностики. Строгие диеты не назначают.
Умеренная ФКУ требует гипофенилаланиновой диеты, а также проведения теста на чувствительность к терапии синтетическим аналогом тетрагидробиоптерина.
Классическая (тяжелая) ФКУ, обусловленная минимальной активностью фермента, лечится строгой гипофенилаланиновой диетой, проводят тест на чувствительность к терапии синтетическим аналогом тетрагидробиоптерина.
Лечение необходимо начинать как можно раньше для предупреждения развития у детей необратимых неврологических нарушений, а у взрослых – когнитивной недостаточности и психических нарушений, вызванных длительным повышением концентрации фенилаланина в крови.
Диетотерапия с жестким ограничением фенилаланина должна быть начата с первых недель жизни ребенка. Недостающее количество белка восполняют за счет лечебных продуктов, частично или полностью лишенных фенилаланина. Из рациона питания больного фенилкетонурией исключают продукты с высоким содержанием белка: мясо, мясопродукты, рыбу, рыбопродукты, творог, яйца, бобовые, орехи, шоколад и др.
В комплекс лечебно-оздоровительных мероприятий, направленных на оптимальное развитие детей с ФКУ входят: медикаментозное лечение, лечебная физкультура (общий массаж, лечебная гимнастика), специализированные педагогические мероприятия.
Осложнения
У детей с фенилкетонурией могут отмечаться аномалии строения черепа, пороки сердца, прогрессирующая умственная отсталость, нарушения речи и памяти, проблемы с концентрацией внимания, расстройства поведения.
Профилактика фенилкетонурии
Основа профилактики – медико-генетическое консультирование пар, планирующих беременность с рекомендацией обследования на гетерозиготное носительство частых мутаций в гене.
Проведение диагностики новорожденных способствует раннему выявлению заболевания и своевременному началу его лечения, что позволяет заметно снизить риск тяжелых проявлений патологии.
Источники:
- Клинические рекомендации «Классическая фенилкетонурия и другие виды гиперфенилаланинемии». Разраб.: Ассоциация медицинских генетиков, Автономная некоммерческая организация «Восточно-Европейская группа по изучению сарком», Союз педиатров России. – 2020.
- Шабалов Н. П. Ш12 Детские болезни: Учебник для вузов. 6-е изд. В двух томах. Т. 2. — СПб.: Питер, 2009. — 928 с.
ВАЖНО!
Информацию из данного раздела нельзя использовать для самодиагностики и самолечения. В случае боли или иного обострения заболевания диагностические исследования должен назначать только лечащий врач. Для постановки диагноза и правильного назначения лечения следует обращаться к Вашему лечащему врачу.
Для корректной оценки результатов ваших анализов в динамике предпочтительно делать исследования в одной и той же лаборатории, так как в разных лабораториях для выполнения одноименных анализов могут применяться разные методы исследования и единицы измерения.
Информация проверена экспертом

Лишова Екатерина Александровна
Высшее медицинское образование, опыт работы — 19 лет
Поделитесь этой статьей сейчас
Рекомендации
-
2477
09 Марта
-
2461
09 Марта
-
2466
07 Марта
Похожие статьи
Нейросифилис
Нейросифилис: причины появления, симптомы, диагностика и способы лечения.
Синдром Дауна
Синдром Дауна: причины появления, симптомы, диагностика и способы лечения.
Гестоз
Гестоз: причины появления, симптомы, диагностика и способы лечения.
Фенилкетонурия
Фенилкетонурия (ФКУ) ― наследственное нарушение метаболизма аминокислот, в первую очередь фенилаланина (ФА), входящего в состав белков. Вещество участвует в укладке белка и стабилизации белковых структур.
Анализы
Первые симптомы: частое срыгивание, рвота, экземы, судороги, исходящий от мочи и кожи запах плесени. Ребенок может был вялым либо, наоборот, гиперактивным. Отстает в психомоторном развитии, наблюдаются признаки олигофрении. Диагноз может быть поставлен в родильном доме. Все дети с фенилкетонурией безусловно получают статус «ребенок-инвалид».
Лечение заболевания заключено в соблюдении специальной низкобелковой диете, не содержащей продукты с ФА.
Определение заболевания
Фенилкетонурия ― это врожденная, генетическая патология, подразумевающая нарушения гидроксилирования фенилаланина. Характеризуется накоплением в организме аминокислоты и продуктов ее метаболизма, что ведет к тяжелым поражениям центральной нервной системы. Впервые заболевание было описано норвежским врачом И. А. Феллингом в 1934 году.
Изучая болезнь специалисты установили, что за наличие болезни отвечает единственный ген фенилаланингидроксилазы. Первое успешное лечение было разработано и проведено в 1950 году в Англии.

В неонатальном периоде клиника отсутствует. Патология проявляется в первые полгода жизни ребенка. В дальнейшем накопление вещества приводит к тяжелым нарушениям развития. Поэтому крайне важно сразу после рождения выявить дефект и не допустить употребление продуктов, содержащих фенилаланин. Более позднее соблюдение диеты не устранит полученные нарушения, но не допустит развития новых.
Патология одинаково часто встречается среди лиц обоих полов. Расовых особенностей не выявлено. Большое количество больных в таких странах как Китай, Турция, Ирландия. В среднем по России с фенилкетонурией рождается каждый 7-ми тысячный ребенок.
Причины фенилкетонурии
Существует три типа генетического отклонения, первый считается классическим, поскольку диагностируется в более чем 90% случаев. Второй и третий ― более редкая форма патологии. Симптоматика схожа во всех типах, заболевание приводит к умственной отсталости. При классической форме фенилкетонурии избежать этого можно диетотерапией, но атипичные варианты, к сожалению, коррекции не подлежат.
- Классическая фенилкетонурия (I тип) ― это низкая выработка фенилаланингидроксилазы (ФАГ), что приводит к собиранию в естественных жидкостях организма фенилаланина и продуктов его расщепления. Патология вызвана мутированным геном РАН.
- Фенилкетонурия II типа ― недостаток дигидроптеридинредуктазы, что препятствует преобразованию фенилаланина в тирозин. Патология из-за мутации гена QDPR.
- Фенилкетонурия III типа ― недостаток 6-пирувоилтетрагидроптеринсинтазы, нужной для синтеза тетрагидробиоптерина. Патология вызвана мутированным PTS-геном.
Все формы заболевания наследуются по аутосомно-рецессивной форме. Это означает, что генетический дефект может быть унаследован у одного из родителей. Половая принадлежность родителя и ребенка не играет роли.

Классификация
Фенилкетонурия в настоящее время не имеет общепризнанной мировой классификации. Над этим вопросом ведутся дебаты, наравне с изучением заболевания. Чуть ранее, до расшифровки генов, считалось, что степень поражения интеллектуальных способностей зависит от степени активности фермента. Поэтому текущая квалификация признана устаревшей. Не учитывает она и другие симптоматические факторы.
При диагностировании ставят:
- I тип (дефицит ФАГ) ― концентрация ФА больше 20 мг/дл.
- Средняя форма ФКУ ― ФА от 8,1 до 20 мг/дл.
- Легкая форма ГФА-уровень ― ФА от 2,1 до 8,0 мг/дл.
При уровне до 8,0 мг/дл фенилкетонурию считают доброкачественной. Она не требует специального лечения, но необходимо наблюдение первый год жизни ребенка. Контролирует состояние врач-педиатр, невролог, генетик.
Выделяют также еще одну форму фенилкетонурии, не требующую коррекции. Это транзиторная форма ГФА в период новорожденности. Возникает, как правило, при недоношенности, что обусловлено функциональной незрелостью организма. Транзиторная фенилкетонурия ― это временное повышение ФА-уровня, способное подняться до критических значений. При этом клиника отсутствует либо проявления весьма незначительны. Через несколько месяцев биохимические показатели приходя в норму.
Патогенез
Механизм зарождения и развития фенилкетонурии связан с нарушением обмена органического соединения ― аминокислоты фенилаланина. Метаболический блок препятствует преобразованию фенилаланина в тирозин. Аминокислота не только не преобразуется, а накапливается в виде токсичных метаболитов:
- фенилмолочная кислота;
- фенилпировиноградная кислота;
- фенилуксусная кислота;
- фенилэтиламин и прочее.
Скопление фенил-веществ оказывает токсическое действие на ЦНС. В настоящий момент механизм еще до конца не изучен, врачам не известен патогенез дисфункции головного мозга.
Существуют предположения, что поражение нервной системы является результатом ряда факторов. Среди них как прямое токсического воздействие фенилаланина, так и нарушение обмена белков, липопротеидов и гликопротеидов, сбой гормонального метаболизма и мембранного транспорта аминокислот. Все это в комплексе имеет важное значение для созревания и правильного функционирования ЦНС.
Симптомы
I тип. Первые признаки у ребенка проявляются в возрасте от 2 месяцев до полугода.
- Апатичность либо, наоборот, повышенная раздражительность.
- Отсутствие интереса к окружению, людям, предметам, обстановке.
- Частое срыгивание.
- Аллергический дерматит.
- Нарушение мышечного тонуса.
- Пониженное давление.
- Судороги.
- Иногда развивается микроцефалия (малый размер черепа относительно других частей тела) и гидроцефалия (избыточная жидкость, омывающая головной мозг).
К характерным симптомам относятся гипопигментация кожи, волос, радужной оболочки глаз. Моча имеет специфический запах плесени или его еще называют «мышиным» запахом. Эпилептические припадки наблюдаются у половины больных, часто является первым выраженным клиническим симптомом. Приступ характеризуется «салаамовыми» судорогами, напоминающими кивки. Они случаются часто, плохо поддаются антиконвульсантному лечению.
Если не корректировать концентрацию ФА, болезнь прогрессирует. Как правило, уровень IQ у таких детей не превышает 20, при норме от 85. Умственная отсталость настолько сильная, что отсутствуют эмоциональные реакции, наблюдаются психопатии и шизофреноподобные расстройства.
II тип. Первая симптоматика проявляется на первом году жизни.
- Повышенная возбудимость.
- Задержка развития.
- Обильное слюнотечение.
- Сниженное артериальное давление.
- Частое повышение температуры тела.
- Сухожильная гиперрефлексия (повышение рефлексов) или спастический тетрапарез (обессиливание всех четырех конечностей).
- Миоклоническая эпилепсия (генерализованные приступы, преимущественно возникающие после пробуждения).
- Микроцефалия.
Отличительная особенность второго типа ― гибель нейронов, нарушение метаболизма фолатов, а также кальцификация в различных отделах головного мозга. Болезнь быстро прогрессирует, может привести к смерти ребенка в течение 2 — 3 лет.
III тип. Симптомы дефицита пирувоилтетрагидроптеринсинтетазы схож с проявлениями болезни Паркинсона:
- Постуральная нестабильность и трудности походки. Сложно либо невозможно поддерживать определенное положение всего тела или конечностей.
- Гипокинезия (низкая двигательная активность, ограниченный темп и объем движений).
- Гиперсаливация (повышенное слюноотделение).
- Нарушения глотания.
- Окулогирные кризы (симметричное отклонение обоих глаз, обычно направленное вверх).
В 80% случаев этот тип заболевания сопровождается снижением количества биогенных аминов в ликворе. Лечение затруднено тем, что раннее снижение концентрации ФА может вызвать серьезные патологические изменения. Несоблюдение диетотерапии приведет к замедлению развития речи, низкому интеллекту, проблемам с памятью.
Диагностика
Выявить фенилкетонурию можно в первые дни после рождения до появления какой-либо симптоматики. Для определение концентрации фениламина в крови проводят:
- микробиологический тест;
- хроматографию;
- флюориметрию;
- масс-спектрометрию.
Во всех случаях биологическим материалом выступают сухие пятна капиллярной крови младенца.

С недавнего времени анализ на фенилкетонурию входит в программу неонатального скрининга. Его проводят всем новорожденным, особенно важно исследование для недоношенных детей. Критерий диагностирования ― повышенная концентрация фенилаланина при норме 0 — 2 мг/дл. Повышенный уровень требует проведения уточняющей диагностики. Потребуется установить сам факт наличия фенилкетонурии и выявить ее причину.
Если скрининг-тест показал высокие результаты уровня ФА, дополнительно может быть назначено:
- Фенилаланин-нагрузочная диагностика для выявления нозологической формы заболевания.
- Молекулярно-генетический анализ для установления формы: классическая, II или III тип.
- Секвенирование гена РАН, если молекулярно-генетическая диагностика дала отрицательный результат по гену ФАГ.
- Анализ на птерины в урине для исключения птерин-зависимых форм.
Дифференциальное диагностирование фенилкетонурии проводят с такими патологиями, как нарушение функции печени, галактоземия и с другими заболеваниями.
Лечение
Симптоматическая терапия при любой формой фенилкетонурии неэффективна. Существует только один способ предотвратить негативные последствия заболевания ― диетотерапия. Из рациона исключают высокобелковые и содержащие фенилаланин продукты. Недостающее количество белка восполняют специализированным лечебным питанием, с максимально низким содержанием аминокислоты ФА или полностью ее лишенным. Следует учитывать, что эффективность терапии напрямую зависит от времени начала коррекции и уже произошедших патологических изменений.
Цель лечебного питания при классической форме заболевания ― это предотвращение развития нарушений ЦНС, физического и умственного развития. Легкая форма ГФА допускает расширение диеты под строгим наблюдением врача за состоянием ребенка и биохимическими показателями. Под запретом: мясо, рыба, орехи, шоколад и бобовые, все виды яиц, творог и сыры. Также следует исключить продукты, содержащие искусственный подсластитель аспартам.
Критерий эффективности лечения ― уровень ФА в крови.
Прогноз и профилактика
Проведения массового скрининга в родильных домах позволяет своевременно выявить генетическое отклонение. Вовремя начать соблюдение диетотерапии и, как следствие, предотвратить тяжелые последствия. В противном случае прогноз в отношении умственного развития неблагоприятный.
Классическая ФКУ имеет благоприятный прогноз если диагностирована в первые недели жизни ребенка и соблюдаются все требования врачей. Такие дети ходят в обычные школы, способны получить высшее образовании и вести нормальный образ жизни.

Во время подготовки к беременности пара должна пройти предварительное генетическое тестирование на наличие мутаций в гене РАН. Если у одного из родителей есть дефектный ген, шанс родить ребенка с ФКУ 1:4 и 100% если оба родителя больны.
Женщины с установленной фенилкетонурией при беременности и грудном вскармливании должны соблюдать строгую диету. Высокая концентрация аминокислоты в крови и околоплодных водах оказывает серьезное тератогенное воздействие на плод.
Преимущества АО «СЗЦДМ»
Сдать анализ на уровень ФА можно в подразделениях АО «СЗЦДМ» Здесь вас ждет:
- квалифицированных и доброжелательный персонал;
- новейшее оборудование, отправка результатов исследований по эл. почте;
- несколько вариантов получения данных анализов;
- удобное расположение терминалов;
- отсутствие очередей, условия конфиденциальности.
Лаборатории находятся в Санкт-Петербурге и других города Ленинградской области, а также в Великом Новгороде, Новгородской обл., Пскове, Калининграде.
Фенилкетонурия и нарушения обмена тетрагидробиоптерина у детей
![]()
Версия: Клинические рекомендации РФ 2013-2017 (Россия)
Категории МКБ:
Другие виды гиперфенилаланинемии (E70.1), Классическая фенилкетонурия (E70.0)
Разделы медицины:
Орфанные заболевания, Педиатрия
Общая информация
Краткое описание
Союз педиатров России
Ассоциация медицинских генетиков
Клинические рекомендации
Фенилкетонурия и нарушения обмена тетрагидробиоптерина у детей
МКБ 10: E70.0/E70.1
Год утверждения (частота пересмотра): 2017(пересмотр каждые 3 года)
Гиперфенилаланинемия (ГФА)
― группа аутосомно-рецессивных заболеваний, обусловленных нарушением обмена незаменимой аминокислоты фенилаланина, поступающей в организм человека с белковой пищей. ГФА объединяет несколько генетически гетерогенных форм нарушения обмена фенилаланина, сходных по клиническим признакам: классическая фенилкетонурия (ФКУ), обусловленная дефицитом фенилаланин-4-гидроксилазы (ФАГ), и гиперфенилаланинемии, связанные с нарушением обмена тетрагидробиоптерина (BH4).
Облачная МИС «МедЭлемент»
Облачная МИС «МедЭлемент»

Автоматизация клиники: быстро и недорого!
- Подключено 300 клиник из 4 стран
- 1 место — 800 RUB / 5500 KZT / 27 BYN в месяц
Классификация
Кодирование по МКБ-10
Е70.0 Классическая фенилкетонурия.
Е70.1 Другие виды гиперфенилаланинемии.
Классификация классической фенилкетонурии основана на степени повышения концентрации фенилаланина в крови, определенной до лечения (на скрининге) (табл. 1). До появления данных молекулярно-генетических исследований ГФА считалось, что тяжесть заболевания и степень поражения интеллекта зависят только от уровня фенилаланина в крови, что тесно связано со степенью активности фермента.
Таблица 1. Рабочая классификация фенилкетонурии, обусловленной дефицитом фермента фенилаланингидроксилазы, в зависимости от уровня фенилаланина крови до лечения [6]
| Форма заболевания | Уровень фенилаланина в сыворотке крови* | |
| мкмоль/л | мг/дл | |
| Легкая ГФА (не ФКУ) | 120−600 | 2−10 |
| Умеренная (мягкая, средняя) ФКУ | 600−1200 | 10−20 |
| Классическая (тяжелая) ФКУ | >1200 | >20 |
Примечание. * ― коэффициент пересчета мкмоль/л в мг/дл равен 60. ГФА ― гиперфенилаланинемия, ФКУ ― фенилкетонурия.
На основе результатов молекулярно-генетических исследований создана современная классификация, представленная в электронной медицинской базе данных «Менделевское наследование у человека» (OMIM), которая хорошо отражает этиопатогенез ГФА и ФКУ (табл. 2).
Таблица 2. Этиопатогенетическая классификация фенилкетонурии и гиперфенилаланинемии [6]
| Код OMIM | Название патологии | Фермент | Ген | Локализация гена |
| 261600 |
ФАГ-зависимая ФКУ (ФКУ, ФАГ-дефицит) |
Фенилаланин-4-гидроксилаза (РАН) | РАН | 12q22-q24.2 |
| 261640 | ГФА, BH4-дефицит, A (ФКУ III типа) | 6-Пирувоил-тетрагидроптерин синтаза (PTPS) | PTS | 11q22.3-q23.3 |
| 233910 | ГФА, BH4-дефицит, B | Гуанозинтрифосфат-циклогидролаза (GTPCH) | GCHI | 14q22.1-q22.2 |
| 261630 | ГФА, BH4-дефицит, C (ФКУ II типа) | Дигидроптеридинредуктаза (DHPR) | QDPR | 4p15.31 |
| 264070 | ГФА, BH4-дефицит, D | Птерин-4-альфа-карбиноламиндегидратаза (PCD) | PCBD | 10q22 |
| 182125 | ГФА, BH4-дефицит | Сепиаптеринредуктаза (SR) | SPR | 1.1.1.153 |
Примечание. ФАГ ― фенилаланингидроксилаза, ГФА ― гиперфенилаланинемия, ФКУ ― фенилкетонурия, BH4 ― тетрагидробиоптерин.
В настоящее время известно несколько форм ВН4-дефицитных ГФА:
• ВН4-дефицитная ГФА (тип А) обусловлена недостаточностью 6-пирувоил-тетрагидроптерин синтазы (PTPS), участвующей в процессе синтеза BH4 из дигидронеоптерин трифосфата. Заболевание вызвано мутацией структурного гена PTS цитозольной 6-PTPS, что приводит к ее недостаточности в печени и эритроцитах. Ген PTS расположен на длинном плече хромосомы 11 в районе q22.3-23.3;
• BH4-дефицитная ГФА (тип В) вследствие недостаточности гуанозинтрифосфат-циклогидролазы I (GTPСH I); кодирующий ген расположен на хромосоме 14q22.2;
• ВН4-дефицитная ГФА (тип С) обусловлена дефицитом дигидроптеридинредуктазы (DHPR), вследствие которого развиваются метаболические блоки на путях превращения фенилаланина в тирозин, а также образования предшественников нейромедиаторов катехоламинового и серотонинового ряда L-дофы и 5-окситриптофана. Заболевание вызвано мутацией структурного гена QDPR цитозольной дигидроптеридинредуктазы. Ген QDPR локализован на хромосоме 4p15.3;
• BH4-дефицитная ГФА (тип D) развивается вследствие недостаточности птерин-4-альфа-карбиноламиндегидратазы (PCВD), которые встречаются реже;
• BH4-дефицитная ГФА вследствие недостаточности сепиаптеринредуктазы (SR).
Тип наследования всех форм ГФА и ФКУ ― аутосомно-рецессивный.
ВН4 является кофактором нескольких важных ферментов, в первую очередь ФАГ, а также тирозингидроксилазы, триптофангидроксилазы и синтазы оксида азота. ВН4-зависимые формы ФКУ имеют сходные клинические проявления с нелеченой классической ФКУ. При этих формах основную роль в патогенезе играет резкая недостаточность нейромедиаторов катехоламинового и серотонинового ряда, поэтому монотерапия диетой не дает положительного результата.
Другие гиперфенилаланинемии могут встречаться при различных физиологических и патологических состояниях.
Транзиторная форма ГФА в период новорожденности ― временное повышение уровня фенилаланина в крови ребенка, в большинстве случаев обусловленное незрелостью ферментативных систем новорожденного (например, при глубокой недоношенности или функциональной незрелости). Провоцирующими факторами развития этого состояния у младенца являются преждевременные роды (вследствие чего не успевает повыситься активность парагидроксифенилпируватоксидазы) и чрезмерное употребление белковой пищи матерью. В результате наблюдается повышенная продукция субстрата, ингибирующего собственный фермент, вследствие чего уровень тирозина и фенилаланина в крови увеличивается до патологических значений. Впоследствии биохимические показатели нормализуются. Клинические проявления либо отсутствуют, либо очень незначительны.
ГФА, сопровождающая поражение печени различной этиологии (вирусное, медикаментозное, токсическое) и другие наследственные нарушения обмена веществ (лейциноз, классическая галактоземия и др.), имеет вторичный характер.
Этиология и патогенез
Фенилкетонурия
(в современной классификации ― ФАГ-зависимая ФКУ) обусловлена дефицитом фермента фенилаланингидроксилазы, приводящим к накоплению в биологических жидкостях фенилаланина (гиперфенилаланинемии) и продуктов его распада. Заболевание вызвано мутацией гена фенилаланингидроксилазы (РАН), локализующегося на длинном плече хромосомы 12, участке 12q22-q24.1.
Нарушения обмена BH4 ― гетерогенная группа гиперфенилаланинемических состояний, вызванная дефицитом одного из ферментов, участвующих в цепи биохимических превращений BH4. Дефицит или недостаточная активность ферментов являются результатом мутаций в соответствующих генах.
В норме в организме человека основное количество фенилаланина утилизируется путем превращения его в тирозин, который в свою очередь служит субстратом для синтеза биогенных аминов и меланина. Лишь небольшое количество фенилаланина используется для синтеза белка. Превращение L-фенилаланина в L-тирозин осуществляется с помощью фермента ФАГ.
В основе патогенеза ГФА лежит блокирование гидроксилирования фенилаланина и превращения его в тирозин. Прямым следствием нарушения гидроксилирования являются накопление фенилаланина в крови и моче и снижение образования тирозина. У нелеченых лиц с ФКУ и ее вариантами, обусловленными недостаточностью BH4, концентрация фенилаланина в плазме достигает уровня, достаточно высокого для активации альтернативных путей метаболизма с образованием фенилпирувата, фенилацетата, фениллактата и других производных, оказывающих токсический эффект на различные органы и ткани (Приложение Г1, рис. 1). В наибольшей степени страдают структуры центральной нервной системы.
Выраженное повреждение мозга может быть связано с рядом эффектов избытка фенилаланина: лишением мозга других аминокислот, необходимых для синтеза белка, что объясняется, вероятно, торможением их всасывания в желудочно-кишечном тракте или нарушением реабсорбции из почечных канальцев в условиях избыточного содержания фенилаланина в жидких средах организма; нарушением образования или стабилизации полирибосом; снижением синтеза миелина и недостаточным синтезом норадреналина и серотонина, имеющих исключительно важную роль в созревании и функционировании центральной нервной системы.
Фенилаланин представляет собой конкурентный ингибитор тирозиназы — ключевого фермента на пути синтеза меланина. Блокада этого пути наряду с уменьшением доступности предшественника меланина (тирозина) обусловливает недостаточную пигментацию волос и кожи.
Активность ФАГ зависит от трех основных кофакторов ― фермента фенилаланингидроксилазы, ВН4, молекулярного кислорода. Наибольшее значение из них имеет BH4. Функция этого кофактора заключается в доставке кислорода к активному центру ФАГ, в котором происходит реакция гидроксилирования фенилаланина. Иными словами, BH4 служит восстановителем для молекулярного кислорода в процессах встраивания его в ряд субстратов (фенилаланин, тирозин, триптофан). Биоптеринзависимыми монооксигеназами также являются тирозиновая и триптофановая гидроксилазы. Реакции, в которых биоптерин играет роль кофактора, представлены в Приложении Г1, рис. 2. В процессе этих реакций кофактор переходит в дигидроформу ― дигидробиоптерин.
Эпидемиология
Частота ФКУ значительно варьирует в зависимости от популяции и составляет от 1:4370 в Турции до 1:80 500 в Японии. Наибольшую распространенность заболевание получило у лиц европеоидной расы, однако и у них частота существенно варьирует в различных регионах и этнических группах. По данным европейских центров неонатального скрининга, частота заболевания в восточно-европейской популяции выше, чем в популяциях запада и юго-запада Европы. Так, частота ФКУ в Ирландии составляет 1:4500 новорожденных, в Югославии ― 1:7300, тогда как в Италии ― 1:12 280, Греции ― 1:18 640. В скандинавских популяциях частота ФКУ исключительно низка, особенно в Финляндии (1:71 000) и Швеции (1:43 230). В России, по данным неонатального скрининга, частота ФКУ составляет 1:7000 и колеблется по регионам от 1:4735 в Курской области до 1:18 000 в Республике Тыва. В Санкт-Петербурге частота ФКУ 1:7600 новорожденных, в Москве ― 1:6772. Наиболее часто встречается классическая форма ФКУ, на долю птеринзависимых форм приходится 1−3% случаев ГФА [1−5].
Фенилкетонурия, обусловленная дефицитом фермента РАН, встречается в большинстве случаев ГФА, выявленных в ходе неонатального скриннга (97−98%).
Гиперфенилаланинемии, связанные с нарушением обмена ВН4 (ранее называемые «атипичная ФКУ»), составляют около 2−3% всех случаев ГФА. Эти состояния обусловлены дефицитом различных ферментов, участвующих в синтезе или восстановлении ВН4.
Диагностика
Жалобы и анамнез
При отсутствии лечения на первом году жизни, обычно в возрасте 2−6 месяцев, родителей беспокоят вялость ребенка, отсутствие интереса к окружающему, иногда повышенная раздражительность, беспокойство, срыгивания, нарушение мышечного тонуса (чаще мышечная гипотония), признаки атопического дерматита, задержка психомоторного развития, иногда судороги. При своевременно назначенном патогенетическом лечении жалобы имеют более легкий характер или отсутствуют (Приложение В).
Физикальное обследование
При отсутствии лечения обращают на себя внимание следующие фенотипические особенности: гипопигментация кожи, волос, радужной оболочки глаз, своеобразный «мышиный» запах мочи больных, возможно формирование микроцефалии. В психоневрологическом статусе отмечаются задержка статико-моторного и психоречевого развития, симптоматическая эпилепсия, а в некоторых случаях ― гидроцефалия.
Эпилептические приступы встречаются почти у половины нелеченых больных и в некоторых случаях могут служить первым признаком болезни. Обычно отмечаются генерализованные пароксизмы по типу инфантильных спазмов в виде «салаамовых судорог», кивков; могут наблюдаться абсансы. Приступы носят упорный характер и плохо поддаются антиконвульсантной терапии. При отсутствии патогенетического лечения болезнь медленно прогрессирует. Умственная отсталость, как правило, достигает глубокой степени: коэффициент умственного развития (intelligence quotient, IQ) составляет около 20 единиц (норма 85−115 единиц). В психологическом статусе больных отмечают нарушение игровой и предметной деятельности, отсутствие дифференцировки эмоциональных реакций, недостаточность экспрессивной и импрессивной речи. Могут наблюдаться двигательные, психопатоподобные и шизофреноподобные расстройства.
Аналогичные клинические симптомы, которые манифестируют после 2 месяцев жизни и достигают максимального проявления к 6 месяцам жизни, имеют BH4-дефицитная ГФА6-PTPS, BH4-дефицитная ГФАGTPSH, BH4-дефицитная ГФА (тип С) вследствие недостаточности DHPR, BH4-дефицитная ГФА вследствие недостаточности SR. Характерны прогрессирующее нарушение психомоторного развития, экстрапирамидные расстройства в виде изменения мышечного тонуса (гипотония мышц туловища и гипертонус мышц конечностей), тремор, атаксия, позднее ― нарушения походки, патологические рефлексы, гиперсаливация, нарушение терморегуляции, псевдобульбарные расстройства в виде затруднения глотания, поперхиваний во время приема пищи, микроцефалия, судороги, окулогирные кризы (эпизодическое содружественное отклонение глаз, обычно направленное вверх и латерально, редко вниз или строго латерально), экзема, гипопигментация кожи. У таких детей при рождении нередко отмечается низкая масса тела.
При недостаточности 6-PTPS различают два фенотипа. Наиболее часто (80%) встречающаяся тяжелая центральная (типичная) форма сопровождается резким дефицитом трансмиттеров и более выраженной тяжестью течения. Вторая, умеренная, периферийная форма сопровождается нормальным содержанием медиаторов, умеренной ГФА и умеренно выраженной клинической симптоматикой.
Для BH4-дефицитной ГФА (тип D) вследствие недостаточности PCВD также характерны специфические изменения мышечного тонуса: постуральная нестабильность, гипокинезия, мышечная дистония (гипертонус конечностей, сниженный тонус мышц туловища).
Лабораторная диагностика
• Рекомендуется проведение неонатального скрининга (определение концентрации фенилаланина в сухих пятнах крови) для доклинической диагностики ГФА и своевременного начала патогенетической терапии [1, 4−7] (Сила рекомендации A; уровень убедительности доказательств II ― здесь и далее Силу рекомендаций и уровни убедительности см. Критерии оценки качества медицинской помощи и Приложение А2).
Комментарии. Все формы ГФА можно диагностировать уже в первые недели или даже дни жизни ребенка, когда клинические проявления еще отсутствуют. Для этого проводят биохимический скрининг новорожденных на наличие ГФА. Подробная схема проведения неонатального скрининга представлена в Приложении Г2, описание правил взятия крови представлено в Приложении Г3.
• Рекомендуется использовать для неонатального скрининга методы флюориметрии или тандемной масс-спектрометрии [1, 4, 6, 7] (Сила рекомендации A; уровень убедительности доказательств II).
Комментарии. Флюориметрия ― количественный биохимический метод определения фенилаланина в крови с помощью современных автоматических флюориметров ― широко используется для проведения массового автоматизированного скрининга. Тандемная масс-спектрометрия ― аналитический метод исследования, основанный на определении отношения массы к заряду ионов, образующихся при ионизации исследуемых компонентов пробы, ― осуществляет качественную и количественную идентификацию аминокислот, позволяет одновременно определять уровни фенилаланина и тирозина, соотношение концентраций позволяет косвенно оценить степень снижения активности ФАГ.
• Главным критерием диагностики ГФА рекомендовано считать повышенное содержание фенилаланина в крови (Сила рекомендации A; уровень убедительности доказательств II).
Комментарии. Для всех указанных методов уровень ФА в крови человека выше 2,0 мг/дл (120 мкмоль/л) квалифицируют как ГФА. ГФА с уровнем выше 10,0 мг/дл (600 мкмоль/л) относят к различным формам ФКУ [6].
• Рекомендуется проведение дифференциальной диагностики ФКУ и других ГФА (второй этап скрининга) [1, 4, 5, 7, 8] (Сила рекомендации A; уровень убедительности доказательств II).
Комментарии. Второй этап скрининга необходим для выявления ВН4-зависимых форм ГФА и своевременного назначения патогенетической терапии. В настоящий момент для этого используется определение соотношения фенилаланин/тирозин (косвенно отражает наличие дефицита фермента РАН) и ДНК-диагностика [1, 7, 9, 10].
• При отсутствии данных неонатального скрининга диагностику заболевания рекомендовано осуществлять на основании совокупности генеалогического анамнеза, результатов клинического и биохимического обследования [1, 6, 8−11] (Сила рекомендации С; уровень убедительности доказательств II).
Комментарии. В этих случаях главным для установления диагноза остается биохимический критерий ― высокое содержание фенилаланина в сыворотке крови при отсутствии патогенетического лечения. В дальнейшем показано проведение молекулярной диагностики.
• Рекомендуется ДНК-диагностика с целью выявления мутаций в генах РАН и РТРS (Сила рекомендации A; уровень убедительности доказательств II).
Комментарии. ДНК-диагностика проводится с целью дифференциальной диагностики и для определения характера мутаций, что в дальнейшем помогает определить контингент пациентов для проведения теста на чувствительность к лечению сапроптерином. Существующие наборы позволяют определять частые мутации в гене РАН, имеющиеся у 80% больных ФКУ. При отсутствии исследуемых мутаций пациенту рекомендуется проведение секвенирования гена РАН. Также проводится ДНК-диагностика мутаций и секвенирование генов (PTPS, DHPR, PCD и др.), кодирующих известные ферменты различных стадий метаболизма BH4.
В настоящее время известно более 900 мутаций в гене РАН, спектр и распространенность которых имеют этнические особенности. Для европеоидной расы мажорной мутацией в гене РАН является мутация R408W, в то время как в Японии и Китае данная мутация не найдена. Во многих европейских популяциях с относительно высокой частотой регистрируются следующие мутации: IVS12nt1, R261Q, R252W, R158Q, P281L, IVS10nt546, I65T.
Большинство генетических изменений гена РАН ― это миссенс-мутации во всех 13 экзонах гена или нетранслируемых фланкирующих участках гена, составляющие 59% всех вариантов. Также обнаружены мутации сплайсинга, нонсенс-мутации, мутации сдвига рамки считывания, более крупные делеции и инсерции. Разные мутации влияют на работу фермента РАН в различной степени: этим может объясняться большое разнообразие показателя фенилаланина в крови больных ФКУ. Большое количество мутаций гена РАН (например, R408W) приводят к резидуальной активности фермента ФАГ. При других мутациях (E390G, Y414C,A300S) толерантность к фенилаланину выше, и клиническая картина ФКУ менее выражена. Таким образом, по современным данным [12], результаты генотипирования при ФКУ потенциально обладают предиктивным значением.
Следует отметить, что при определенных мутациях в гене РАН, обусловливающих остаточную активность фермента ФАГ, введение кофактора BH4 или его синтетических аналогов в терапию приводит к повышению или восстановлению активности ФАГ, что позволяет расширить диету с увеличением в рационе квоты натурального белка.
Инструментальная диагностика
• Рекомендовано проведение электроэнцефалографии для выявления паттернов гипсаритмии (даже при отсутствии клинических судорожных приступов), единичных и множественных спайк- и полиспайкразрядов, других изменений [1, 13] (Сила рекомендации С; уровень убедительности доказательств II).
• Рекомендовано проведение магнитно-резонансной томографии с целью выявления очагов перивентрикулярной лейкопатии, кортикальной атрофии и других изменений у пациентов старше 12 лет [1, 14, 15] (Сила рекомендации С; уровень убедительности доказательств II).
• Рекомендовано проведение ультразвукового исследования брюшной полости и почек для диагностики дискинезии желчных путей, диффузных изменений печени и поджелудочной железы, мочекаменной болезни [1, 7, 16] (Сила рекомендации С; уровень убедительности доказательств II).
• Рекомендовано проведение эзофагогастрофиброскопии для диагностики поражения слизистой оболочки желудка (по показаниям) [1, 7, 15, 16] (Сила рекомендации С; уровень убедительности доказательств II).
Иная диагностика (консультативная помощь)
• Рекомендовано психолого-педагогическое консультирование и логопедическое тестирование [1, 7, 13, 17] (Сила рекомендации С; уровень убедительности доказательств II).
Комментарии. Тестирование проводится с целью определения уровня интеллектуального и речевого развития, возможностей социальной адаптации и составления индивидуального плана психолого-педагогического сопровождения.
Рекомендовано проведение по показаниям консультаций специалистов (невропатолога, гастроэнтеролога, офтальмолога, аллерголога и др.) [7, 16, 18] (Сила рекомендации С; уровень убедительности доказательств II).
Дифференциальный диагноз
Дифференциальная диагностика
• Рекомендовано проводить дифференциальную диагностику с целью выявления ВН4-зависимых форм ГФА и определения потенциально чувствительных больных с РАН-зависимой ГФА (фенилкетонурией) (Сила рекомендации С; уровень убедительности доказательств II).
• Рекомендовано после подтверждения у новорожденного РАН-зависимой ГФА провести тест на потенциальную чувствительность к сапроптерина дигидрохлориду, после чего назначают лечение [11, 19, 20] (Сила рекомендации С; уровень убедительности доказательств II).
Комментарий. Процедура проведения тестирования описана в Приложении Г3. Уровень фенилаланина в крови до начала тестирования должен быть ≥450 мкмоль/л. Дополнительную информацию для дифференциальной диагностики можно получить при исследовании птеринов мочи, сыворотки крови и спинномозговой жидкости (в Российской Федерации не проводится) (табл. 3).
Таблица 3. Диагностические показатели фенилананина и птеринов в крови, моче и спинномозговой жидкости [8]*

Примечание. * ― AdGTPSH ― аутосомно-доминантный дефицит гуанозин-трифосфат-циклогидролазы I, ArGTPSH ― аутосомно-рецессивный дефицит гуанозин-трифосфат-циклогидролазы I, PTPS ― 6-пирувоил-тетрагидроптерин синтаза, SR ― сепиаптеринредуктаза, PCD ― птерин-4-альфа-карбиноламиндегидратаза, DHPR ― дигидроптеридинредуктаза, Phe ― фенилаланин, Neo ― неоптерин, Bio ― биоптерин, Pri ― примаптерин, SHIAA ― S-гидроксииндолуксусная кислота, HVA ― гомованилиновая кислота, SMTHF ― 5-метитетрагидрофолат, К ― кровь, М ― моча, СМЖ ― спинномозговая жидкость; n ― норма, ↑/↓ ― повышение/понижение значения показателя.
Лечение
Основная цель лечения — снизить фенилаланин в крови, повысить толерантность (переносимость) фенилаланина, получаемого с натуральной пищей, и таким образом избежать тяжелой неврологической симптоматики и улучшить качество жизни.
Подходы к терапии при различной тяжести течения гиперфенилаланинемии
Легкая ГФА требует наблюдения и проведения дифференциальной диагностики. Строгого диетического лечения при этой форме ГФА, как правило, не назначают, хотя в последние годы рекомендуют начинать лечение при уровне фенилаланина в крови ≥360 мкмоль/л [8]. Дети с легкой формой ГФА должны находиться под систематическим наблюдением врача в течение первого года жизни с контролем уровня фенилаланина в крови, проведением необходимых диагностических мероприятий с целью исключения птеринзависимых форм ГФА и выбора дальнейшей тактики лечения.
Умеренная (мягкая, средняя) ФКУ подразумевает сохранение частичной активности фермента ФАГ, требует назначения гипофенилаланиновой диеты, а также проведения теста на чувствительность к терапии синтетическим аналогом BH4.
Классическая (тяжелая) ФКУ обусловлена минимальной активностью фермента, требует назначения строгой гипофенилаланиновой диеты, а также проведения теста на чувствительность к терапии синтетическим аналогом BH4.
Патогенетически обоснованной терапией для больных с ГФА, обусловленной недостаточностью BH4, является назначение синтетического аналога ВН4 ― сапроптерина дигидрохлорида, который используется в комплексе с диетотерапией или без нее (в зависимости от формы болезни) и симптоматической медикаментозной терапией.
Консервативное лечение
• Рекомендуется назначение патогенетически обоснованной для РАН-зависимой ФКУ и ГФА гипофенилаланиновой диеты [7, 15, 21, 22] (Сила рекомендации С; уровень убедительности доказательств II).
Комментарии. Диетотерапия, основанная на резком ограничении фенилаланина в рационе больных детей за счет исключения высокобелковых продуктов, должна быть начата не позднее первых недель жизни ребенка с целью достижения максимальной эффективности лечения. Недостающее количество белка восполняется за счет специализированных лечебных продуктов, частично или полностью лишенных фенилаланина.
Согласно современным данным, диетическое лечение назначают при уровне фенилаланина на скрининге ≥360 мкмоль/л (≥6 мг/дл).
Из рациона питания больного ФКУ исключают продукты с высоким содержанием белка (соответственно, и фенилаланина) ― мясо, мясопродукты, рыбу, рыбопродукты, творог, яйцо, бобовые, орехи, шоколад и др. Допустимые в диете натуральные продукты, такие как женское молоко, детские молочные смеси (для детей в возрасте до 1 года), овощи, фрукты и некоторые другие продукты с низким содержанием белка вводят в соответствии с подсчетом содержащегося в них фенилаланина.
• При расчетах питания рекомендуется ориентироваться на нормы физиологической потребности в основных нутриентах для больных детей различных возрастных групп в соответствии с МР 2.3.1.2432-08 [22] (табл. 4, 5); допускается уменьшение количества суточного белка (не более 10%) в зависимости от толерантности больного к пище и к фенилаланину, а также от состояния нутритивного статуса (Сила рекомендации С; уровень убедительности доказательств II).
Таблица 4. Среднесуточные нормы потребностей в основных пищевых веществах и энергии для детей первого года жизни (на кг массы тела) [22]
| Возраст, мес | Энергия, ккал/ кг | Белок, г/кг | Жиры, г/кг | Углеводы, г/кг |
| 0−3 | 115 | 2,2 | 6,5 | 13 |
| 4−6 | 115 | 2,6 | 6,0 | 13 |
| 7−12 | 110 | 2,9 | 5,5 | 13 |
Таблица 5. Нормы физиологической потребности в основных пищевых веществах и энергии для детей старше 1 года [22]
| Возраст, лет | Энергия, ккал | Белок, г/день* | Жиры, г/день | Углеводы, г/день |
| От 1 до 2 | 1200 | 36 (28) | 40 | 174 |
| От 2 до 3 | 1400 | 42 (33) | 47 | 203 |
| От 3 до 7 | 1800 | 54 (46) | 60 | 261 |
| От 7 до 11 | 2100 | 63 (54) | 70 | 305 |
| От 11 до 14 мальчики | 2500 | 75 (64) | 83 | 363 |
| От 11 до 14 девочки | 2300 | 69 (59) | 77 | 334 |
| От 14 до 18 юноши | 2900 | 87 (74 | 97 | 421 |
| От 14 до 18 девушки | 2500 | 76 (64) | 83 | 363 |
Примечание. * ― в скобках указано ориентировочное потребление белка за счет специализированной смеси без фенилаланина.
Комментарии. Белок за счет естественных продуктов в диете рассчитывается исходя из допустимых суточных количеств фенилаланина с учетом, что 1 г белка содержит ~50 мг фенилаланина. В зависимости от переносимости пищевого фенилаланина допустимое и безопасное его количество в сутки составляет от 90 до 35 мг/кг массы тела для детей первого года жизни. В питании детей старше 1 года допустимое количество фенилаланина постепенно снижается с 35 до 10 мг/кг массы тела ребенка (табл. 6).
Таблица 6. Допустимое количество фенилаланина в питании детей с фенилкетонурией в зависимости от возраста
| Возраст детей | Количество фенилаланина (мг/кг массы тела в сутки) |
| от 0 до 2 мес | 90−60 |
| 2−6 мес | 55−45 |
| 6−12 мес | 40−35 |
| 1−3 года | 35−25 |
| 3−7 лет | 25−20 |
| 7 лет и старше | 20−10 |
Недостающее количество белка восполняется за счет специализированных лечебных продуктов ― смесей аминокислот без фенилаланина и низкобелковых продуктов питания. Аминокислотные смеси различаются по содержанию белка (от 13 г до 77,5 г на 100 г сухого продукта) и других питательных веществ (углеводы, жиры, витамины, микро- и макроэлементы). Все смеси в своем составе не содержат фенилаланина. Аминокислотные смеси с содержанием 13−15 г белка в 100 г сухой смеси предназначены для детей первого года жизни. Детям более старшего возраста назначаются смеси с более высоким содержанием белка (Приложение Г4).
• На первом году жизни необходимы ежедневный подсчет количества фенилаланина, получаемого с пищей, употребляемой пациентом, учет белков, жиров, углеводов, энергии в суточном рационе (Сила рекомендации С; уровень убедительности доказательств II).
Комментарии. Расчет суточной дозы специализированного продукта производится по формуле:
(Ps-Pn) × 100 /P
где Ps ― суточное количество белка, Pn ― белок естественных продуктов, P ― количество белка в 100 г сухого специализированного продукта.
Пример расчета питания ребенку с ФКУ
Пациент, возраст 3 года, масса тела 14,5 кг
1. Общее суточное количество белка в рационе больного (см. табл. 5) ― 54,0 г.
2. Допустимое суточное количество фенилаланина (см. табл. 6): 25×14,5=363 мг.
3. Допустимое количество белка естественных продуктов (1 г белка содержит 50 мг фенилаланина): 363:50=7,3 г.
4. Количество белка за счет специализированного продукта на основе смеси L-аминокислот без фенилаланина: 54,0-7,3=46,7 г.
5. Суточное количество аминокислотной смеси (100 г содержит 20 г белка): 46,7×100:20=233 г.
6. Рекомендуемое суточное количество жира в рационе (см. табл. 5) ― 60 г.
7. Рекомендуемое суточное количество углеводов (см. табл. 5) ― 261 г.
• При организации диетотерапии рекомендовано учитывать клиническую форму заболевания, уровень фенилаланина в крови, возраст ребенка; нутритивный статус (физическое развитие), толерантность ребенка к пищевому фенилаланину, количество фенилаланина и натурального белка, получаемого с пищей, количество основных пищевых веществ и энергии в лечебном рационе [7, 15, 21, 22] (Сила рекомендации С; уровень убедительности доказательств II).
Комментарии. При назначении диеты важен индивидуальный и дифференцированный подход к использованию специализированных и натуральных продуктов соответственно возрасту ребенка.
Для больных ФКУ независимо от их возраста сохраняется запрет на продукты, наиболее богатые фенилаланином, такие как мясо, рыба и изделия из них. Творог, твердые сыры, бобовые, куриные яйца, орехи могут в ограниченном количестве входить в рацион пациентов старшего возраста с учетом толерантности к фенилаланину. Не рекомендуется употребление пациентами с ФКУ продуктов фастфуд, газированных напитков с подсластителями (аспартам или пищевая добавка Е951), содержащих фенилаланин.
Тактика диетотерапии при сопутствующих заболеваниях (выраженная гипертермия, интоксикация, различные диспепсические явления), а также при отказе от приема аминокислотной смеси заключается в кратковременном (на 2−3 дня) прекращении диетотерапии с заменой лечебных продуктов на натуральные с невысоким содержанием белка. После стихания острого периода болезни в рацион ребенка вновь вводится специализированный продукт, но за более короткий период, чем в начале лечения. Если ребенок с ФКУ не отказывается от пищи во время болезни, то лечение сопутствующих соматических проводится по общепринятой схеме и не требует прекращения диетотерапии.
• Рекомендуется (по показаниям) медикаментозная терапия сапроптерином ФКУ, обусловленной дефектами обмена ФАГ (легкие и умеренные формы), после проведения теста и подтверждения чувствительности к сапроптерину [2, 6, 8, 9, 15] (Сила рекомендации С; уровень убедительности доказательств II).
Комментарии. Синтетический аналог ВН4 ― сапроптерина дигидрохлорид ― является патогенетическим методом лечения для ВН4-дефицитных форм ГФА и вспомогательным методом лечения чувствительных к ВН4 терапии форм классической ФКУ.
Тестирование потенциальной чувствительности к лечению и лечение сапроптерином проводит и контролирует врач, который осуществляет также и наблюдение пациентов с ФКУ (Приложение Г5).
Ответ на лечение препаратом сапроптерина дигидрохлоридом оценивается по степени снижения концентрации фенилаланина в крови больного при соблюдении стабильной гипофенилаланиновой диеты. Пациент считается чувствительным, если разница уровня фенилаланина, полученного по окончании периода оценки ответа на лечение, и его исходного уровня перед началом приема препарата составляет 30% и более.
При необходимости длительная медикаментозная терапия у больных ФКУ, отвечающих на лечение сапроптерина дигидрохлоридом снижением уровня фенилаланина в крови, проводится в комбинации с диетой при использовании аминокислотных смесей, количество которых определяет врач.
• Для ВН4-дефицитных форм ГФА в качестве патогенетического метода лечения рекомендуется применение синтетического аналога ВН4 ― сапроптерина дигидрохлоридаж (А16АХ) в сочетании с гипофенилаланиновой диетой (табл. 7) (Сила рекомендации C; уровень убедительности доказательств II).
Комментарии. Начальная доза сапроптерина дигидрохлорида у больных с недостаточностью BH4 составляет от 2 до 5 мг/кг массы тела при приеме 1 раз в день. Доза может быть увеличена до 20 мг/кг массы тела в день. Для достижения оптимального терапевтического эффекта суточная доза препарата может быть разделена на 2 или 3 приема в течение дня.
• В комплекс лечения также рекомендовано включать препараты леводопыж,вк(10−15 мг/кг в сутки) в сочетании с карбидопойж,вк(Код АТХ: N04BA) в дозировке 1−1,5 мг/кг в сутки, 5-гидрокситриптофан (10 мг/кг в сутки) (препарат в Российской Федерации в настоящее время не зарегистрирован), 5-формилтетрагидрофолат (кальция фолинатж,вк Код АТХ: V03AF) в средней дозе 25 мг/сут, в некоторых случаях диета с ограничением фенилаланина и фолиевой кислотыж,вк(Код АТХ: B03BB) (см. табл. 7).
Таблица 7. Схема терапии при различных ВН4-дефицитных состояниях (с/без гиперфенилаланинемии)*

Примечание. * ― таблица составлена по данным [8], ** ― препарат не зарегистрирован в Российской Федерации.
Иное лечение
Хирургического вмешательства обычно не требуется.
Реабилитационные мероприятия направлены на улучшение когнитивных и речевых функций, психоэмоционального состояния, социальной адаптации.
Прогноз
Прогноз заболевания зависит от своевременной диагностики, максимально раннего назначения диетотерапии и адекватного ее выполнения и контроля. Большое значение имеет приверженность лечению всех членов семьи больного, а в дальнейшем ― и самого пациента, для чего необходимо организовать психолого-педагогическую поддержку, которая должна начинаться с момента рождения больного ребенка.
В случае несоблюдения рекомендаций по диетотерапии и недостаточном контроле за уровнем фенилаланина в крови могут иметь место такие отдаленные последствия, как сниженный коэффициент интеллекта, нарушения речи и памяти, проблемы с концентрацией внимания, расстройства поведения.
В случае если пациенты не принимают специализированные аминокислотные смеси без фенилаланина, а находятся на диете с резким ограничением высокобелковых продуктов, возможно развитие симптомов хронической недостаточности питания, нутритивного дефицита витаминов, макро- и микроэлементов и других эссенциальных факторов.
Для классической ФКУ, выявленной в первые недели жизни ребенка, прогноз при соблюдении рекомендаций врачей по лечению по заболеванию благоприятный. Дети посещают массовые детские и образовательные учреждения, занимаются в дополнительных кружках, в дальнейшем поступают в высшие учебные заведения.
Профилактика
Профилактика и диспансерное наблюдение
Профилактика ФКУ и ГФА проводится в нескольких направлениях:
• проспективное медико-генетическое консультирование пар, планирующих беременность, с рекомендацией обследования на гетерозиготное носительство частых мутаций в гене РАН. При выявлении ФКУ в семье ― обследование родственников для уточнения гетерозиготного носительства мутации;
• в семье, где имеется ребенок с ФКУ, при последующей беременности ― проведение пренатальной диагностики с целью определения наличия патологии у плода;
• проведение неонатального скрининга с охватом 100% новорожденных, позволяющего своевременно выявить заболевание и приступить к лечению, чтобы избежать тяжелых проявлений патологии;
• профилактика рождения детей с синдромом «материнской фенилкетонурии» от женщин, больных ФКУ, путем организации психологической помощи девочкам-подросткам по вопросам необходимости соблюдения строгой гипофенилаланиновой диеты в пубертатный период, а также консультативной помощи по вопросам планирования семьи и беременности.
Диспансерное наблюдение
Заболевание диагностируется в результате неонатального скрининга, выявленные больные находятся на диспансерном учете в медико-генетических центрах (консультациях), где назначается лечение, включающее диетотерапию, и осуществляется контроль за адекватностью его проведения.
Рекомендуется следующая схема контроля за содержанием фенилаланина в крови у больных ФКУ:
• в возрасте до 3 месяцев ― 1 раз в неделю (до получения стабильных результатов) и далее 1 раз в 10 дней;
• с 1 года до 6 лет ― не менее 2 раз в месяц;
• с 7 до 12 лет ― не менее 1 раза в месяц;
• после 12 лет ― 1 раз в 2 месяца.
Терапевтический диапазон уровня фенилаланина в сыворотке крови может быть расширен в зависимости от возраста и ослабления диетических ограничений (табл. 8).
Таблица 8. Рекомендуемый уровень фенилаланина в сыворотке крови у больных фенилкетонурией, находящихся на лечении
| Возраст, лет | Уровень фенилаланина | |
| мкмоль/л | мг/дл | |
| 0−6 | 120−360 | 2−6 |
| 7-9 | 120−360 | 2−6 |
| 10−12 | 120−360 | 2−6 |
| 13-15 | 120−600 | 2−10 |
| 16−18 | 120−900 | 2−15 |
На фоне лечения необходимо проводить контроль за нутритивным статусом больного, физическим и интеллектуальным развитием. Большинство детей с ФКУ находятся на этапе амбулаторно-поликлинического наблюдения, где осуществляется контроль за состоянием их здоровья с привлечением врачей-специалистов, использованием функциональных методов исследования (УЗИ, ЭЭГ, МРТ), а также контроль клинико-лабораторных показателей (общие анализы крови и мочи, общий белок и его фракции, по показаниям липидный профиль, глюкоза, ферритин, креатинин, сывороточное железо и др.); 1 раз в год рекомендуется исследовать аминокислотный спектр крови. Общий анализ крови рекомендуется делать не реже 1 раза в 6 месяцев, биохимический ― 1 раз в год. Консультация психолога для больных ФКУ подросткового возраста является обязательной и должна проходить ежегодно. Контроль фосфорно-кальциевого обмена (кальций, фосфор, остеокальцин, паратгормон и др.) должен проводиться с одного года жизни. Пациентам старше 13 лет рекомендуется проведение денситометрии 1 раз в год.
Девочкам с ФКУ независимо от возраста рекомендуется поддерживать содержание фенилаланина в крови на уровне до 4 мг/дл (240 мкмоль/л) с целью профилактики в дальнейшем синдрома «материнской фенилкетонурии» у будущего потомства [26−28].
Информация
Источники и литература
-
Клинические рекомендации Союза педиатров России
- 1. Бушуева Т.В. Современный взгляд на проблему фенилкетонурии у детей: диагностика, клиника, лечение. Вопросы современной педиатрии. 2010;9(11):57−162.
2. Бушуева Т.В., Кузенкова Л.М., Боровик Т.Э., Назаренко Л.П., и др. Открытое несравнительное клиническое исследование III фазы по оценке частоты ответа и безопасности сапроптерина у пациентов с фенилкетонурией и гиперфенилаланинемией. Вестник Российской академии медицинских наук. 2014;7–8:69−77.
3. Матулевич С.А. Компьютеризация и программное обеспечение неонатального скрининга на наследственные болезни обмена. Медицинская генетика. 2009;8;3(81):35−38.
4. Матулевич С.А., Голихина Т.А. Неонатальный скрининг на наследственные болезни / Наследственные болезни: национальное руководство. Глава 32 / Под ред. акад. РАМН Н.П. Бочкова, акад. РАМН Е.К. Гинтера, акад. РАМН В.П. Пузырева. М.:ГЭОТАР-Медиа; 2012. 936 с.
5. Cunningham A, Bausell H, Brown M, et al. Recommendations for the use of sapropterin in phenylketonuria. Mol Genet Metab. 2012;106:269–276.
6. Bushueva TV, Vinyarskaya IV, Chernikov VV, Borovik TE, Kuzenkova LM. Quality of life in russian children with phenylketonuria. J Inher Metab Dis. 2014 Sept;37(Suppl 1):S60−61.
7. Баранов А.А., Боровик Т.Э., Ладодо К.С., Бушуева Т.В., Маслова О.И., Кузенкова Л.М., Студеникин В.М., Звонкова Н.Г. и др. Специализированные продукты лечебного питания для детей с фенилкетонурией. Методическое письмо. 3-е издание. Москва; 2012. 84 с.
8. Blau N, Burton BK, Thony B, van Spronsen FJ, Waisbren S. Phenylketonuria and BH4 Deficiencies. UNI-MED Verlag AG. Bremen-London-Boston; 2014. 79 p.
9. Гундорова П., Степанова А.А., Щагина О.А., Поляков А.В. Результаты использования новых медицинских технологий «Детекция основных точковых мутаций гена РАН методом мультиплексной лигазной реакции» и «Детекция десяти дополнительных точковых мутаций гена РАН методом мультиплексной лигазной реакции» в ДНК диагностике фенилкетонурии. Медицинская генетика. 2016;15;2(164):29−36.
10. Burton BK, Grange DK, Milanowski A, et al. The response of patients with phenylketonuria and elevated serum phenylalanine to treatment with oral sapropterin dihydrochloride (6R-tetrahydrobiopterin): a phase II, multicentre, open-label, screening study. J Inherit Metab Dis. 2007;30:700–707.
11. Howell RR. National Institutes of Health Consensus Development Conference Statement: phenylketonuria: screening and management, October 16–18, 2000. Pediatrics. 2001;108(4):972–982.
12. Trunzo R, Santacroce R, Shen N, Jung-Klawitter S, Leccese A, De Girolamo G, Margaglione M, Blau N.In vitro residual activity of phenylalanine hydroxylase variants and correlation with metabolic phenotypes in PKU. Gene. 2016 Dec 5;594(1):138-143.
13. Gordon P, Thomas JA, Suter R, Jurecki E. Evolving patient selection and clinical benefit criteria for sapropterin dihydrochloride (Kuvan) treatment of PKU patients. Mol Genet Metab. 2012;105:672–676.
14. Ng TW, Rae A, Wright H, Gurry D, Wray J. Maternal phenylketonuria in Western Australia: pregnancy outcomes and developmental outcomes in offspring. J Paediatr Child Health. 2003;39:358–363.
15. Боровик Т.Э., Ладодо К.С., Грибакин С.Г., Скворцова В.А., Бушуева Т.В., Вознесенская Т.С., Заплатников А.Л., Захарова И.Н., Звонкова Н.Г., Картамышева Н.Н., Коровина Н.А., Кураева Т.Л., Кутафина Е.К., Мазанкова Л.Н., Макарова С.Г., Мухина Ю.Г., Нетребенко О.К., Потапов А.С., Рославцева Е.А., Рыбакова Е.П., и др.
Клиническая диетология детского возраста. Руководство для врачей / Под редакцией Т.Э. Боровик, К.С. Ладодо. Москва: Издательство «Медицинское информационное агентство»; 2008. 614 с.
16. Голихина Т.А., Люманова Э.Р. Психологический статус личности детей с фенилкетонурией, получающих диетотерапию с раннего возраста. Кубанский научный медицинский вестник. 2011;8(122):50−53.
17. Ладодо К.С., Рыбакова Е.П., Соломадина Л.В. Специализированное лечебное питание для детей с фенилкетонурией. Руководство по фармакотерапии в педиатрии и детской хирургии. Клиническая генетика / Под ред. А.Д. Царегородцева, В.А. Таболина. Москва; 2002. С. 132−138.
18. Blau N, Hennermann JB, Langenbeck U, Lichter-Konecki U. Diagnosis, classification, and genetics of phenylketonuria and etrahydrobiopterin (BH4) deficiencies. Mol Genet Metab. 2011;104:S2–S9.
19. Chace DH, Millington DS, Terada N, Kahler SG, Roe CR, Hofman LF. Rapid diagnosis of phenylketonuria by quantitative nalysis or phenylalanine and tyrosine in neonatal blood spots by tandem mass spectrometry. Clin Chem. 1993;39:66–71.
20. Боровик Т.Э., Ладодо К.С., Бушуева Т.В. и др. Диетотерапия при классической фенилкетонурии: критерии выбора специализированных продуктов без фенилаланина. Вопросы современной педиатрии. 2013;12(5):40–48.
21. van Spronsen FJ, van Wegberg AMJ, Ahring K, Belandger-Quitana A, Blau N, Bosch AM, et al. European Guidelines on diagnosis and treatment of PKU. J Inherit Metab Dis. 2016;39(Suppl):150. URL: http://www.ssiem2016.org/wp/wp-content/uploads/2016/09/SSIEM-2016-content-pdf.pdf
22. MP 2.3.1.2432-08 Нормы физиологических потребностей в энергии и пищевых веществах для различных групп населения Российской Федерации (утв. Главным государственным санитарным врачом РФ 18 декабря 2008 г.).
23. Singh RH, Rohr F, Frazier D, et al. Recommendations for the nutrition management of phenylalanine hydroxylase deficiency. Genet Med. January, 2; 2014.
24. Anjema K, van Rijn M, Hofstede FC, Bosch AM, Hollak CE, Rubio-Gozalbo E, de Vries MC, Janssen MC, Boelen CC, Burgerhof JG, Blau N, Heiner-Fokkema MR, van Spronsen FJ. Tetrahydrobiopterin responsiveness in phenylketonuria: prediction with the 48-hour loading test and genotype. Orphanet J Rare Dis. 2013 Jul 10;8:103.
25. Burton BK, Grange DK, Milanowski A, Vockley G, Feillet F, Crombez EA, Abadie V, Harding CO, Cederbaum S, Dobbelaere D, Smith A, Dorenbaum A. The response of patients with phenylketonuria and elevated serum phenylalanine to treatment with oral sapropterin dihydrochloride (6R-tetrahydrobiopterin): a phase II, multicentre, open-label, screening study. J Inherit Metab Dis. 2007 Oct;30(5):700−7.
26. Денисенкова Е.В., Кузнецова Л.И. Влияние материнской фенилкетонурии на исход беременности и родов. Вопросы детской диетологии. 2009;7(3):55−59.
27. Levy HL, Milanowski A, Chakrapani A, et al. Sapropterin Research Group (Efficacy of sapropterin dihydrochloride tetrahydrobiopterin, 6R-BH4) for reduction of phenylalanine concentration in patients with phenylketonuria: a phase III randomised placebo-controlled study. Lancet. 2007;370:504–510.
28. Lee PJ, Ridout D, Walter JH, Cockburn F. Maternal phenylketonuria: report from the United Kingdom Registry 1978−97. Arch Dis Child. 2005;90:143–146.
- 1. Бушуева Т.В. Современный взгляд на проблему фенилкетонурии у детей: диагностика, клиника, лечение. Вопросы современной педиатрии. 2010;9(11):57−162.
Информация
Ключевые слова
Гиперфенилаланинемия
Гипофенилаланиновая диета
Классическая фенилкетонурия
Сапроптерина дигидрохлорид
Тетрагидробиоптерин
Тирозин
Фенилаланин
Фенилаланин-4-гидроксилаза
Список сокращений
ГФА ― гиперфенилаланинемия
МГК ― медико-генетическая консультация
МРТ ― магнитно-резонансная томография
УЗИ ― ультразвуковое исследование
ФАГ ― фермент фенилаланингидроксилаза
ФКУ ― фенилкетонурия
ЭЭГ ― электроэнцефалография
AdGTPCH (autosomal dominant guanosine triphosphate cyclohydrolase) ― аутосомно-доминантный дефицит гуанозинтрифосфат-циклогидролазы I
ArGTPCH (autosomal recessive guanosine triphosphate cyclohydrolase) ― аутосомно-рецессивный дефицит гуанозинтрифосфат-циклогидролазы I
ВН4 (tetrahydrobiopterin) ― кофактор тетрагидробиоптерин
DHPR (dihydropteridine reductase) ― фермент дигидроптеридинредуктаза
GTPСH (guanosine triphosphate cyclohydrolase) ― фермент гуанозинтрифосфатциклогидролаза
OMIM (online mendelian inheritance of man) ― электронная база данных «Менделевское наследование у человека»
PAH (phenylalanine hydroxylase) ― фермент фенилаланингидроксилаза
PCBD (pterin-4-alpha-carbinolamine dehydratase) ― фермент птерин-4-альфа-карбиноламиндегидратаза
PTPS (6-pyruvoyl tetrahydropterin synthase) ― фермент 6-пирувоил-тетрагидроптерин синтаза
SR (sepiapterin reductase) ― фермент сепиаптеринредуктаза
Термины и определения
«Материнская фенилкетонурия» ― эмбриофетопатия, развивающаяся у плода в результате воздействия продуктов аномального метаболизма беременной женщины с фенилкетонурией при отсутствии диетического лечения.
Неонатальный скрининг ― медицинская диагностическая технология сплошного безвыборочного лабораторного обследования всех новорожденных на некоторые заболевания обмена веществ, призванная обеспечить своевременное выявление и начало лечения больных детей с целью предотвращения их инвалидизации.
Пренатальная диагностика фенилкетонурии ― комплексная дородовая диагностика с целью выявления фенилкетонурии на стадии внутриутробного развития путем определения активности фенилаланингидроксилазы в культуре амниоцитов, биоптате и культуре хориона.
Критерии оценки качества медицинской помощи
Организационно-технические условия оказания медицинской помощи
| Вид медицинской помощи | Специализированная, в том числе высокотехнологичная, медицинская помощь |
| Возрастная группа | Дети |
| Условия оказания медицинской помощи | Стационарно, в дневном стационаре |
| Форма оказания медицинской помощи | Плановая |
Критерии оценки качества медицинской помощи
| № | Критерии качества | Сила рекомендаций | Уровень убедительности доказательств |
| 1 | Выполнен неонатальный скрининг | А | I |
| 2 | Выполнено определение уровня аминокислот и органических кислот в сыворотке крови и патологических метаболитов в моче методом тандемной масс-спектрометрии | А | I |
| 3 | Выполнено определение чувствительности к сапроптерину | С | II |
| 4 | Проведена гипофенилаланиновая диета под контролем фенилаланина крови и коррекцией не реже 1 раза в 3 месяца | С | II |
| 5 | Проведена терапия сапроптерином при ВН4-чувствительной форме (при отсутствии противопоказаний) | С | II |
| 6 | Выполнен контроль фенилаланина крови не реже 1 раза в 3 месяца | С | II |
Приложение А1. Состав рабочей группы
ФГАУ «ННПЦЗД» Минздрава России
А.А. Баранов ― академик РАН, д.м.н., профессор, член Исполкома Союза педиатров России
Л.С. Намазова-Баранова ― академик РАН, д.м.н., проф., Председатель Исполкома Союза педиатров России
К.С. Ладодо ― д.м.н., проф., член Союза педиатров России
Т.Э. Боровик ― д.м.н., проф., член Союза педиатров России
Т.В. Бушуева ― д.м.н., член Союза педиатров России
Л.М. Кузенкова ― д.м.н., проф., член Союза педиатров России
К.В. Савостьянов — к.б.н., член Союза педиатров России
Е.А. Вишнёва ― к.м.н., член Союза педиатров России
Н.В. Журкова ― к.м.н., член Союза педиатров России
Н.Г. Звонкова ― к.м.н., член Союза педиатров России
Л.Р. Селимзянова ― к.м.н., член Союза педиатров России
А.А. Пушков ― к.б.н., член Союза педиатров России
ФГБОУ ВО РНИМУ им. Н.И. Пирогова Минздрава России
П.В. Новиков ― д.м.н., проф.
Е.А. Николаева ― д.м.н., проф.
ФГБНУ «Медико-генетический научный центр»
С.И. Куцев ― д.м.н., проф., член-корр. РАН, президент Ассоциации медицинских генетиков
Е.С.Тюменцева – д.м.н., член Ассоциации медицинских генетиков
А.В. Поляков ― д.б.н., проф., член Ассоциации медицинских генетиков
Е.Ю. Захарова ― д.м.н., проф., член Ассоциации медицинских генетиков
П. Гундарова, член Ассоциации медицинских генетиков
Московский центр неонатального скрининга
Е.А. Шестопалова, член Ассоциации медицинских генетиков
ГБУЗ «Краевая клиническая больница №1 имени проф. С.В.Очаповского» МЗ Краснодарского края. Кубанская межрегиональная медико-генетическая консультация
С.А. Матулевич ― д.м.н., член Ассоциации медицинских генетиков
Т.А. Голихина ― к.м.н., член Ассоциации медицинских генетиков
ГБУЗ г.Москвы Научно-практический центр психического здоровья детей и подростков
Е.В. Денисенкова ― к.м.н.
СПб ГКУЗ «Диагностический центр (медико-генетический)»
О.П. Романенко ― д.м.н., проф., член Ассоциации медицинских генетиков
Л.В. Лязина ― к.м.н., член Ассоциации медицинских генетиков
НИИ медицинской генетики ФГБНУ «Томский национальный исследовательский медицинский центр Российской академии наук»
Л.П. Назаренко ― д.м.н., проф., член Ассоциации медицинских генетиков
АНМО «Ставропольский краевой клинический консультативно-диагностический центр» медико-генетическая консультация
Е.Г. Бакулина ― к.м.н., член Ассоциации медицинских генетиков
Консультативно-диагностический центр «Охрана здоровья матери и ребенка» г. Екатеринбург
В.К. Подолина, член Ассоциации медицинских генетиков
Е.Б. Николаева, заслуженный врач РФ
Н.В. Никитина, к.м.н., член Ассоциации медицинских генетиков
Приложение А2. Методология разработки клинических рекомендаций
Целевая аудитория данных клинических рекомендаций:
• врачи-педиатры (код специальности 31.05.02);
• врачи общей семейной практики (семейная медицина) (код специальности 31.08.54);
• генетики (код специальности 31.08.30);
• диетологи (код специальности 31.08.34);
• невропатологи (код специальности 31.08.42);
• медицинские психологи (код специальности 19.00.04);
• дефектологи (код специальности 44.03.03).
Таблица П1. Уровни убедительности доказательств
| Уровни убедительности доказательств | Источник доказательств |
| I (1) |
Проспективные рандомизированные контролируемые исследования. Достаточное количество исследований с достаточной мощностью, с участием большого количества пациентов и получением большого количества данных. Крупные метаанализы. Как минимум одно хорошо организованное рандомизированное контролируемое исследование. Репрезентативная выборка пациентов |
| II (2) |
Проспективные с рандомизацией или без исследования с ограниченным количеством данных. Несколько исследований с небольшим количеством пациентов. Хорошо организованное проспективное исследование когорты. Метаанализы ограничены, но проведены на хорошем уровне. Результаты непрезентативны в отношении целевой популяции. Хорошо организованные исследования «случай-контроль» |
| III (3) |
Нерандомизированные контролируемые исследования. Исследования с недостаточным контролем. Рандомизированные клинические исследования как минимум с 1 значительной или как минимум 3 незначительными методологическими ошибками. Ретроспективные или наблюдательные исследования. Серия клинических наблюдений. Противоречивые данные, не позволяющие сформировать окончательную рекомендацию |
| IV (4) | Мнение эксперта / данные из отчета экспертной комиссии, экспериментально подтвержденные и теоретически обоснованные |
Таблица П2. Сила рекомендаций
| Сила рекомендаций | Описание | Расшифровка |
| А | Рекомендация основана на высоком уровне доказательности (как минимум 1 убедительная публикация I уровня доказательности, показывающая значительное превосходство пользы над риском) | Метод/терапия первой линии; либо в сочетании со стандартной методикой/терапией |
| В | Рекомендация основана на среднем уровне доказательности (как минимум 1 убедительная публикация II уровня доказательности, показывающая значительное превосходство пользы над риском) | Метод/терапия второй линии; либо при отказе, противопоказании или неэффективности стандартной методики/терапии. Рекомендуется мониторирование побочных явлений |
| С |
Рекомендация основана на слабом уровне доказательности (но как минимум 1 убедительная публикация III уровня доказательности, показывающая значительное превосходство пользы над риском) или нет убедительных данных ни о пользе, ни о риске) |
Нет возражений против данного метода/терапии или нет возражений против продолжения данного метода/терапии. Рекомендовано при отказе, противопоказании или неэффективности стандартной методики/терапии при условии отсутствия побочных эффектов |
| D | Отсутствие убедительных публикаций I, II или III уровня доказательности, показывающих значительное превосходство пользы над риском, либо убедительные публикации I, II или III уровня доказательности, показывающие значительное превосходство риска над пользой | Не рекомендовано |
Приложение А3. Связанные документы
1. Приказ Минздравсоцразвития РФ от 22.03.2006 г. № 185 «О массовом обследовании новорожденных детей на наследственные заболевания».
2. Приказ Министерства здравоохранения и социального развития РФ от 16 апреля 2012 г. № 366н «Об утверждении Порядка оказания педиатрической помощи».
3. Приказ Министерства здравоохранения РФ от 15 ноября 2012 г. № 917н «Об утверждении Порядка оказания медицинской помощи больным с врожденными и (или) наследственными заболеваниями».
4. Постановление Правительства Российской Федерации от 9 апреля 2015 г. № 333 «Об утверждении Правил формирования перечня специализированных продуктов лечебного питания для детей-инвалидов».
Приложение Б Алгоритмы ведения пациента

Приложение В Информация для пациентов
Фенилкетонурия ― это наследственное заболевание, характеризующееся нарушением метаболизма фенилаланина (незаменимая аминокислота, которая не синтезируется в организме, поступает с пищей ― продуктами животного происхождения, в том числе с грудным молоком и детскими молочными смесями).
При отсутствии своевременной диагностики и лечения заболевание проявляется обычно в возрасте 2−6 месяцев жизни признаками поражения центральной нервной системы: родителей беспокоят вялость ребенка, отсутствие интереса к окружающему, иногда повышенная раздражительность, беспокойство, срыгивания, нарушение мышечного тонуса (чаще мышечная гипотония), признаки атопического дерматита, задержка психомоторного развития, иногда судороги. С возрастом дети имеют тяжелое поражение нервной системы вплоть до умственной отсталости и эпилепсии. При своевременно назначенном патогенетическом лечении жалобы имеют более легкий характер или отсутствуют.
С целью ранней диагностики и профилактики развития указанных симптомов проводится неонатальный скрининг, результаты которого сообщаются родителям по телефону.
Родители обязаны внимательно отнестись к полученной из центра неонатального скрининга информации и немедленно явиться на прием к врачу и/или связаться с ним по телефону, даже если у ребенка отсутствуют клинические симптомы. Решение о назначении и характере диетотерапии принимает врач.
Диетотерапия, основанная на резком ограничении фенилаланина в рационе больных детей за счет исключения высокобелковых продуктов, должна быть начата не позднее первых недель жизни ребенка с целью достижения максимальной эффективности лечения. Недостающее количество белка восполняется за счет специализированных лечебных продуктов, частично или полностью лишенных фенилаланина.
Назначение патогенетической диетотерапии с первых дней жизни ребенка определяет благоприятный прогноз течения фенилкетонурии.
Учитывая аутосомно-рецессивный тип наследования и высокий риск повторного рождения в семье больного ребенка, показано медико-генетическое консультирование семей.
Приложение Г1 Схема метаболических процессов, приводящих к развитию гиперфенилаланинемии (фенилкетонурии)

Рис. 1. Реакция 1 ― преобразование фенилаланина в тирозин под действием фермента фенилаланингидроксилазы; реакция 2 – тетрагидробиоптерин в присутствии Fe2+ под действием фермента дигидробиоптеринредуктазы окисляется до образования дигидробиоптерина

Рис. 2. Метаболизм тетрагидробиоптерина
Приложение Г2 Схема неонатального скрининга на фенилкетонурию

Примечание. МГК ― медико-генетическая консультация, ФА ― фенилаланин, ГФА ― гиперфенилаланинемия, ФКУ ― фенилкетонурия.
Приложение Г3 Правила взятия крови у новорожденного
Взятие крови у новорожденного проводится на 4−5-й день жизни (у недоношенных детей ― на 7-й день жизни), но не ранее чем через 2 дня от начала энтерального питания. Взятие крови ранее 4 дней жизни нежелателен из-за большого числа ложноположительных и ложноотрицательных результатов тестирования.
Образец крови берут из пятки новорожденного ребенка через 3 часа после кормления. Предварительно необходимо согреть стопу ребенка, обернув ее полотенцем, смоченным теплой водой (не выше 42°С) на 1−2 минуты, затем протереть область пункции стерильной салфеткой, смоченной 70% спиртом. Во избежание гемолиза крови обработанное место следует промокнуть сухой стерильной салфеткой.
Место прокола расположено медиально от линии, проведенной от большого пальца до пятки, или латерально от линии, проведенной от мизинца до пятки (заштрихованная область на рис.). Глубина пунктирования не должна превышать 2−5 мм, т.к. при более глубоком проколе возникает опасность остеомиелита.

Рис. Взятие образца крови из пятки новорожденного
Прокол осуществляется одноразовым скарификатором, первая капля крови снимается стерильным сухим тампоном. Вторая капля крови наносится на тест-бланк, пропитываемый кровью полностью и насквозь в указанных местах (кружки). Чрезмерное сдавливание места прокола может вызвать гемолиз или примешивание к образцу тканевой жидкости.
Кровь наносится только на лицевую сторону бланка. На каждую область кровь наносится только один раз. Запрещено наслаивать на уже нанесенную кровь второй слой, это искажает результаты исследования. Если крови мало, лучше правильно заполнить меньше областей, чем все, но неправильно.
В связи с тем, что скрининг проводится на несколько болезней обмена, пятен должно быть не менее пяти. Вид пятен должен быть одинаковым с обеих сторон тест-бланка.
Бланк с кровью высушивается в горизонтальном положении на чистой обезжиренной поверхности в течение 3 часов при комнатной температуре (15−22°С). Не допускается соприкосновение бланков между собой во время сушки. Нельзя использовать любые виды нагрева бланка (солнечный свет, фен, батарея и т.п.) для ускорения сушки.
В обменной карте новорожденного делается отметка о проведенном заборе крови. В случае отсутствия в документации новорожденного ребенка отметки о заборе образца крови при его поступлении под наблюдение в детскую поликлинику по месту жительства или переводе по медицинским показаниям в больничное учреждение забор образца крови осуществляется в указанных медицинских учреждениях в соответствии с рекомендациями. Высушенный тест-бланк с образцом крови отправляется с соблюдением температурного режима в лабораторию неонатального скрининга.
Неправильная подготовка пациента к взятию крови, нарушение техники взятия пробы, правил хранения и транспортировки биологического материала, может привести к ошибочным результатам лабораторных исследований. Ложноположительные результаты скрининга приводят к увеличению количества необходимых повторных исследований и расхода реактивов, неоправданному психологическому стрессу у родителей. Ложноотрицательные результаты могут привести к пропуску скринируемого заболевания. В процессе наблюдения за ребенком педиатр может выявить клинические симптомы, характерные для наследственных болезней обмена, даже при отрицательных результатах скрининга. В этом случае следует провести углубленное обследование ребенка.
Приложение Г4 Химический состав специализированных продуктов лечебного питания для больных фенилкетонурией
(в 100 г сухого продукта, для жидких продуктов указано отдельно)
| Наименование продукта | Белок (эквивалент), г | Жир, г | Углеводы, г | Энергетическая ценность, ккал |
| Продукты для детей первого года жизни | ||||
| Афенилак 131 | 13 | 25 | 52 | 485 |
| Афенилак 151 | 15 | 23 | 51,7 | 474 |
|
Нутриген 14 ― phe PREMIUM1 |
14 | 23 в т.ч. ДЦПНЖК | 52 | 460 |
| Анамикс ХР LCP | ||||
| МD мил ФКУ-0 | 13 | 23 в т.ч. ДЦПНЖК | 59 | 495 |
| COMIDA-PKU А формула + LCP | 11,8 | 27,4 в т.ч. ДЦПНЖК | 52,6 | 506 |
| Продукты для больных фенилкетонурией старше 1 года | ||||
| Афенилак 201 | 20 | 18 | 50,4 | 444 |
| Афенилак 401 | 40 | 13,5 | 31 | 405 |
| Нутриген 301 | 30 | 0 | 54 | 336 |
| Нутриген 701, 2 | 70 | 0 | 6,9 | 308 |
| Нутриген 751, 2 | 75 | 0 | 1,3 | 305 |
|
Нутриген 20 ― phe PREMIUM1 |
20 | 18 | 48 | 434 |
|
Нутриген 40 ― phe PREMIUM1 |
40 | 13,5 | 28 | 393 |
|
Нутриген 70 ― phe PREMIUM1, 2 |
70 | 0 | 0-12 | 280 |
| П-АМ 1 | 75 | 0 | 0 | 300 |
| П-АМ 2 | 75 | 0 | 0 | 300 |
| П-АМ 32 | 75 | 0 | 0 | 300 |
| ХР Максамейд | 39 | Менее 0,5 | 34 | 297 |
| ХР Максамум | 25 | Менее 0,5 | 51 | 309 |
| Изифен (100 мл) | 6,7 | 2,0 | 5,1 | 65 |
| Лофлекс (62,5мл) | 10 | — | 4,4 | 58 |
| П-АМ материнский2 | 77,5 | 0 | 0 | 310 |
| PKU Nutri 2 Energy | 27 | 14 | 42 | 402 |
| МD мил ФКУ-1 | 20 | 0 | 73 | 372 |
| МD мил ФКУ-2 | 40 | 6,1 | 46,9 | 402 |
| МD мил ФКУ-32 | 69,1 | 0 | 23 | 368 |
| МD мил ФКУ Премиум2 | 69,1 | 1,9 | 23 | 385 |
| COMIDA-PKU В формула | 31,1 | 15 | 40,6 | 422 |
| COMIDA-PKU В формула Клубника | 31,1 | 14,2 | 40,5 | 419 |
| COMIDA-PKU В формула Карамель | 31,1 | 14,8 | 40,6 | 422 |
| COMIDA-PKU В формула Шоколад | 31,1 | 14,2 | 38,7 | 415 |
| COMIDA-PKU В формула Апельсин-Лимон | 31,1 | 13,1 | 41,1 | 412 |
| COMIDA-PKU В2 | 73 | 0 | 0,5 | 296 |
| COMIDA-PKU С формула | 45 | 0 | 38,9 | 335 |
| COMIDA-PKU С формула Апельсин-Лимон | 45 | 0 | 36 | 331 |
| COMIDA-PKU С2 | 75 | 0 | 0,4 | 302 |
| COMIDA-PKU С2 капсулы | 75 | 0 | 0,4 | 302 |
Примечание. 1 ― специализированные продукты отечественного производства; 2 ― специализированные продукты, которые могут использоваться в диетотерапии женщин с фенилкетонурией в период преконцепционной подготовки и во время беременности. ДЦПНЖК ― длинноцепочечные полиненасыщенные жирные кислоты.
Приложение Г5 Тест на чувствительность к сапроптерину дигидрохлориду
Тестирование потенциальной чувствительности к сапроптерину и дальнейшее лечение им проводит и контролирует врач, который осуществляет наблюдение пациентов с ФКУ. Для более тщательного контроля тест целесообразно проводить в стационаре, возможно дневного пребывания, при удовлетворительной комплаентности пациента ― амбулаторно [1, 2, 11, 19]. Перед проведением тестирования у родителей/законных представителей ребенка, а также у пациентов старше 14 лет берут информированное согласие на проведение данной процедуры.
Цель тестирования ― выявить ВН4-зависимые формы ГФА и определить чувствительность к сапроптерину у больных с классической ФКУ.
Тестирование возможно проводить по 2-дневной или 7-дневной схеме [23, 24].
Дозировка препарата во время тестирования составляет от 5 до 20 мг/кг массы тела (в среднем 10 мг/кг массы тела); некоторые авторы рекомендуют максимальную дозировку (20 мг/кг) для проведения тестирования [24]. Суточная доза рассчитывается индивидуально для каждого пациента в соответствии с инструкцией к препарату.
Перед проведением тестирования стабилизируют содержание фенилаланина в крови путем поддержания постоянства пищевого рациона в течение 4−5 дней до начала и в период тестирования.
Тестирование начинают на фоне стабильного уровня фенилаланина крови, при этом рассчитывается среднее значение двух показателей фенилаланина, полученных в результате двукратного взятия крови перед началом тестирования: целесообразно, чтобы среднее значение фенилаланина было не ниже 450 мкмоль/л (7,5 мг%) [23−25].
В день первого приема препарата натощак берут анализ крови из пальца на фенилаланин, затем ребенок принимает рассчитанную дозу препарата вместе с завтраком или сразу же после завтрака. В течение 2 (или 7) дней утром в одно и то же время после завтрака (или вместе с едой) ребенок принимает препарат, разведенный в теплой воде в соответствии с инструкцией.
Повторное взятие крови проводят на 3-й (или 8-й) день строго натощак. Ответ на чувствительность к препарату оценивается по степени снижения концентрации фенилаланина в крови больного при соблюдении стабильной гипофенилаланиновой диеты. Пациент считается чувствительным, если разница уровня фенилаланина, полученного по окончании периода оценки ответа на лечение, и его исходного уровня перед началом приема препарата составляет 30% и более.
При снижении уровня фенилаланина более 85% у пациента высока вероятностьдиагноза ГФА, обусловленной недостаточностью ВН4.
Важным условием тестирования является стабильная гипофенилаланиновая диета с целью исключения ее влияния на колебания концентрации фенилаланина в крови.
Схемы тестирования приведены в табл. 1, 2.
Таблица 1. Двухдневная схема тестирования для определения чувствительности к сапроптерину

Таблица 2. Семидневная схема тестирования для определения чувствительности к сапроптерину

Повышение толерантности к пищевому фенилаланину на фоне приема сапроптерина является показанием для расширения диеты, при этом концентрация фенилаланина в крови не должна превышать целевого значения 6 мг% (360 мкмоль/л). При комбинированном использовании гипофенилаланиновой диеты и сапроптерина контроль за антропометрическими и биохимическими показателями нутритивного статуса должен быть систематическим во избежание развития дефицита основных и минорных пищевых веществ, алиментарно-зависимых состояний.
При необходимости длительная медикаментозная терапия у больных ФКУ, отвечающих на лечение сапроптерина дигидрохлоридом снижением уровня фенилаланина в крови, проводится в комбинации с диетой при использовании аминокислотных смесей, количество которых определяет врач.
После проведения тестирования и получения положительного результата необходимо подтвердить его в ходе длительного наблюдения. Пациенты, получающие сапроптерин, требуют тщательного контроля состояния их нутритивного статуса и других показателей здоровья не реже 1 раза в 6 месяцев.
Медикаментозная терапия при ВН4-дефицитных состояниях с/без ГФА: начальная доза сапроптерина дигидрохлорида у больных с недостаточностью BH4 составляет от 2 до 5 мг/кг массы тела при приеме 1 раз в день. При необходимости доза сапроптерином может быть увеличена до 20 мг/кг массы тела в день, для достижения оптимального терапевтического эффекта возможен прием препарата 2−3 раза в день.
Приложение Г6 Расшифровка примечаний
…ж ― лекарственный препарат, входящий в Перечень жизненно необходимых и важнейших лекарственных препаратов для медицинского применения на 2017 год (Распоряжение Правительства РФ от 28 декабря 2016 года N 2885-р);
…вк —лекарственный препарат, входящий в Перечень лекарственных препаратов для медицинского применения, в том числе лекарственных препаратов для медицинского применения, назначаемых по решению врачебных комиссий медицинских организаций (Распоряжение Правительства РФ от 26.12.2015 N 2724-р).
Прикреплённые файлы
Мобильное приложение «MedElement»

- Профессиональные медицинские справочники. Стандарты лечения
- Коммуникация с пациентами: онлайн-консультация, отзывы, запись на приём
Скачать приложение для ANDROID / для iOS
Мобильное приложение «MedElement»

- Профессиональные медицинские справочники
- Коммуникация с пациентами: онлайн-консультация, отзывы, запись на приём
Скачать приложение для ANDROID / для iOS
Внимание!
Если вы не являетесь медицинским специалистом:
-
Занимаясь самолечением, вы можете нанести непоправимый вред своему здоровью.
-
Информация, размещенная на сайте MedElement и в мобильных приложениях «MedElement (МедЭлемент)», «Lekar Pro»,
«Dariger Pro», «Заболевания: справочник терапевта», не может и не должна заменять очную консультацию врача.
Обязательно
обращайтесь в медицинские учреждения при наличии каких-либо заболеваний или беспокоящих вас симптомов.
-
Выбор лекарственных средств и их дозировки, должен быть оговорен со специалистом. Только врач может
назначить
нужное лекарство и его дозировку с учетом заболевания и состояния организма больного.
-
Сайт MedElement и мобильные приложения «MedElement (МедЭлемент)», «Lekar Pro»,
«Dariger Pro», «Заболевания: справочник терапевта» являются исключительно информационно-справочными ресурсами.
Информация, размещенная на данном
сайте, не должна использоваться для самовольного изменения предписаний врача.
-
Редакция MedElement не несет ответственности за какой-либо ущерб здоровью или материальный ущерб, возникший
в
результате использования данного сайта.
Фенилкетонурия – это наследственное нарушение аминокислотного обмена, обусловленное недостаточностью печеночных ферментов, участвующих в метаболизме фенилаланина до тирозина.
В среднем фенилкетонурии подвержен 1 из 8000 человек.
Симптомы
Фенилкетонурия проявляется на первом году жизни. Основными симптомами в этом возрасте являются:
* вялость ребенка;
* отсутствие интереса к окружающему;
* иногда повышенная раздражительность;
* беспокойство;
* срыгивания;
* нарушения мышечного тонуса (чаще мышечная гипотония);
* судороги;
* признаки аллергического дерматита;
* появляется характерный «мышиный» запах мочи.
В более позднем возрасте для больных фенилкетонурией характерна задержка психоречевого развития, нередко отмечается микроцефалия.
При фенилкетонурии характерны следующие фенотипические особенности: гипопигментация кожи, волос, радужной оболочки глаз. У некоторых больных одним из проявлений патологии может быть склеродермия.
Эпилептические приступы встречаются почти у половины больных фенилкетонурией и в некоторых случаях могут служить первым признаком болезни.
Диагностика
В настоящее время диагностика фенилкетонурии (а также галактоземии, врожденного гипотиреоза, адрено-генитального синдрома и муковисцидоза) входит в программу неонатального скрининга, осуществляемого всем новорожденным. Основные и дополнительные методы диагностики:
* Скрининг-тест. Проводится на 3-5 день жизни доношенного и 7 день жизни недоношенного ребенка путем забора образца капиллярной крови на специальный бумажный бланк. При обнаружении гиперфенилаланемии более 2,2 мг% ребенка направляют к детскому генетику для повторного обследования.
* Биохимические исследования. Для подтверждения диагноза фенилкетонурии проверяется концентрация фенилаланина и тирозина в крови, определяют активность печеночных ферментов (фенилаланингидроксилазы), выполняется биохимическое исследование мочи (определение кетоновых кислот), метаболитов катехоламинов в моче и др.
* Неврологическое обследование. Дополнительно проводится ЭЭГ и МРТ головного мозга, осмотр ребенка детским неврологом.
* Пренатальная диагностика. Генетический дефект при фенилкетонурии может быть обнаружен еще на этапе беременности в ходе инвазивной пренатальной диагностики плода (хорионбиопсии, амниоцентеза, кордоцентеза).
Лечение
Основополагающим фактором в лечении фенилкетонурии является соблюдение диеты, ограничивающей поступление белка в организм. Лечение рекомендуется начинать при концентрации фенилаланина >6 мг%. Для грудных детей разработаны специальные смеси — Афенилак, Лофенилак; для детей старше 1 года – Тетрафен, Фенил-фри; старше 8 лет — Максамум-ХР и др. Основу диеты составляют низкобелковые продукты — фрукты, овощи, соки, белковые гидролизаты и аминокислотные смеси. Расширение диеты возможно после 18 лет в связи с возрастанием толерантности к фенилаланину. В соответствии с российским законодательством обеспечение лиц, страдающих фенилкетонурией, лечебным питанием, должна осуществляться бесплатно.
Больным назначается прием минеральных соединений, витаминов группы В и др.; по показаниям — ноотропные средства, антиконвульсанты. В комплексной терапии фенилкетонурии широко используется общий массаж, ЛФК. Атипичные формы фенилкетонурии, не поддающиеся лечению диетой, требуют назначения гепатопротекторов, противосудорожных средств, заместительной терапии леводопой, 5-гидрокситриптофаном.
Куван — препарат, который является аналогом естественного кофермента тетрагидробиоптерина или ВН4, который вырабатывается в организме. Мутации в гене фенилаланингидроксилазы и выработка в организме «неактивных» белковых молекул ФАГ являются основной причиной развития фенилкетонурии. Действие препарата заключается в активации поврежденных молекул ФАГ, которые начинают обеспечивать превращение фенилаланина в тирозин более эффективно. Таким образом, применение Кувана позволяет контролировать уровень фенилаланина даже при существенном расширении диеты. Особенно препарат эффективен при умеренных и мягких формах ФКУ и у некоторых больных позволяет полностью обходиться без диеты.
Дети, страдающие фенилкетонурией, находятся под наблюдением участкового педиатра и психоневролога; нередко нуждаются в помощи логопеда и дефектолога. Необходим тщательный мониторинг нервно-психического статуса детей, контроль уровня фенилаланина в крови и показателей электроэнцефалограммы.
Прогноз и профилактика
Проведения массового скрининга на фенилкетонурию в неонатальном периоде позволяет организовать раннюю диетотерапию и предотвратить тяжелые церебральные повреждения, нарушения функции печени. При раннем назначении элиминационной диеты при классической фенилкетонурии прогноз развития детей хороший. При поздно начатом лечении прогноз в отношении умственного развития неблагоприятный.
Профилактика осложнений фенилкетонурии заключается в проведении массового скрининга новорожденных, раннего назначения и длительного соблюдения диетического питания.